Guidance Document
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Antimicrobial Activity of Cationic
ANTIMICROBIAL ACTIVITY OF CATIONIC ANTISEPTICS IN LAYER-BY-LAYER THIN FILM ASSEMBLIES A Thesis by CHARLENE MYRIAH DVORACEK Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE May 2009 Major Subject: Mechanical Engineering ANTIMICROBIAL ACTIVITY OF CATIONIC ANTISEPTICS IN LAYER-BY-LAYER THIN FILM ASSEMBLIES A Thesis by CHARLENE MYRIAH DVORACEK Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Approved by: Chair of Committee, Jaime Grunlan Committee Members, Michael Benedik Xinghang Zhang Head of Department, Dennis O'Neal May 2009 Major Subject: Mechanical Engineering iii ABSTRACT Antimicrobial Activity of Cationic Antiseptics in Layer-by-Layer Thin Film Assemblies. (May 2009) Charlene Myriah Dvoracek, B.S., Rose-Hulman Institute of Technology Chair of Advisory Committee: Dr. Jaime Grunlan Layer-by-layer (LbL) assembly has proven to be a powerful technique for assembling thin films with a variety of properties including electrochromic, molecular sensing, oxygen barrier, and antimicrobial. LbL involves the deposition of alternating cationic and anionic ingredients from solution, utilizing the electrostatic charges to develop multilayer films. The present work incorporates cationic antimicrobial agents into the positively-charged layers of LbL assemblies. When these thin films are exposed to a humid environment, the antimicrobial molecules readily diffuse out and prevent bacterial growth. The influence of exposure time, testing temperature, secondary ingredients and number of bilayers on antimicrobial efficacy is evaluated here. Additionally, film growth and microstructure are analyzed to better understand the behavior of these films. -

Reseptregisteret 2013–2017 the Norwegian Prescription Database
LEGEMIDDELSTATISTIKK 2018:2 Reseptregisteret 2013–2017 Tema: Legemidler og eldre The Norwegian Prescription Database 2013–2017 Topic: Drug use in the elderly Reseptregisteret 2013–2017 Tema: Legemidler og eldre The Norwegian Prescription Database 2013–2017 Topic: Drug use in the elderly Christian Berg Hege Salvesen Blix Olaug Fenne Kari Furu Vidar Hjellvik Kari Jansdotter Husabø Irene Litleskare Marit Rønning Solveig Sakshaug Randi Selmer Anne-Johanne Søgaard Sissel Torheim Utgitt av Folkehelseinstituttet/Published by Norwegian Institute of Public Health Område for Helsedata og digitalisering Avdeling for Legemiddelstatistikk Juni 2018 Tittel/Title: Legemiddelstatistikk 2018:2 Reseptregisteret 2013–2017 / The Norwegian Prescription Database 2013–2017 Forfattere/Authors: Christian Berg, redaktør/editor Hege Salvesen Blix Olaug Fenne Kari Furu Vidar Hjellvik Kari Jansdotter Husabø Irene Litleskare Marit Rønning Solveig Sakshaug Randi Selmer Anne-Johanne Søgaard Sissel Torheim Acknowledgement: Julie D. W. Johansen (English text) Bestilling/Order: Rapporten kan lastes ned som pdf på Folkehelseinstituttets nettsider: www.fhi.no The report can be downloaded from www.fhi.no Grafisk design omslag: Fete Typer Ombrekking: Houston911 Kontaktinformasjon/Contact information: Folkehelseinstituttet/Norwegian Institute of Public Health Postboks 222 Skøyen N-0213 Oslo Tel: +47 21 07 70 00 ISSN: 1890-9647 ISBN: 978-82-8082-926-9 Sitering/Citation: Berg, C (red), Reseptregisteret 2013–2017 [The Norwegian Prescription Database 2013–2017] Legemiddelstatistikk 2018:2, Oslo, Norge: Folkehelseinstituttet, 2018. Tidligere utgaver / Previous editions: 2008: Reseptregisteret 2004–2007 / The Norwegian Prescription Database 2004–2007 2009: Legemiddelstatistikk 2009:2: Reseptregisteret 2004–2008 / The Norwegian Prescription Database 2004–2008 2010: Legemiddelstatistikk 2010:2: Reseptregisteret 2005–2009. Tema: Vanedannende legemidler / The Norwegian Prescription Database 2005–2009. -

Being Aware of Chlorhexidine Allergy
Being aware of chlorhexidine allergy If you have an immediate allergic reaction to chlorhexidine you may experience symptoms such as: x itching x skin rash (hives) x swelling x anaphylaxis. People who develop anaphylaxis to chlorhexidine may have experienced mild reactions, such as skin rash, to chlorhexidine before. Irritant contact dermatitis or allergic contact dermatitis Chlorhexidine can also cause irritant dermatitis. This is not a true allergic reaction. It is caused by chlorhexidine directly irritating skin and results in rough, dry and scaly Chlorhexidine is an antiseptic. Allergic reactions to skin, sometimes with weeping sores. chlorhexidine are rare but are becoming more common. Chlorhexidine is used in many products both in Chlorhexidine can also cause allergic contact hospitals and in the community. dermatitis. Symptoms look like irritant dermatitis, but the cause of the symptoms is delayed by 12-48 hours Why have I been given this factsheet? after contact with chlorhexidine. You have been given this brochure because you have had a reaction to a medication, a medical dressing Both irritant dermatitis and allergic contact dermatitis or antiseptic. This may or may not be caused by a caused by chlorhexidine are annoying but not chlorhexidine allergy. dangerous. It is important that you are aware of the possibility of an It is recommended that you avoid chlorhexidine if you allergy. experience these responses as some people have gone on to develop immediate allergic reaction to chlorhexidine. Allergic reactions to chlorhexidine Severe allergic reactions to chlorhexidine are rare, but How do I know which products contain they can be serious. Immediate allergic reactions can chlorhexidine? cause anaphlaxis (a very severe allergic reaction which can be life-threatening). -

National OTC Medicines List
National OTC Medicines List ‐ DraŌ 01 DRAFT National OTC Medicines List Draft 01 Ministry of Public Health of Lebanon This list was prepared under the guidance of His Excellency Minister Waêl Abou Faour andDRAFT the supervision of the Director General Dr. Walid Ammar. Editors Rita KARAM, Pharm D. PhD. Myriam WATFA, Pharm D Ghassan HAMADEH, MD.CPE FOREWORD According to the French National Agency for Medicines and Health Products Safety (ANSM), Over-the-counter (OTC) drugs are medicines that are accessible to patients in pharmacies, based on criteria set to safeguard patients’ safety. Due to their therapeutic class, these medicines could be dispensed without physician’s intervention for diagnostic, treatment initiation or maintenance purposes. Moreover, their dosage, treatment period and Package Insert Leaflet should be suitable for OTC classification. The packaging size should be in accordance with the dosage and treatment period. According to ArticleDRAFT 43 of the Law No.367 issued in 1994 related to the pharmacy practice, and the amendment of Articles 46 and 47 by Law No.91 issued in 2010, pharmacists do not have the right to dispense any medicine that is not requested by a unified prescription, unless the medicine is mentioned in a list which is established by pharmacists and physicians’ syndicates. In this regard, the Ministry of Public Health (MoPH) developed the National OTC Medicines List, and presentedit in a scientific, objective, reliable, and accessible listing. The OTC List was developed by a team of pharmacists and physicians from the Ministry of Public Health (MoPH). In order to ensure a safe and effective self- medicationat the pharmacy level, several pharmaceutical categories (e.g. -

List Item Cetrimide: Summary Report
The European Agency for the Evaluation of Medicinal Products Veterinary Medicines Evaluation Unit EMEA/MRL/073/96-FINAL March 1996 COMMITTEE FOR VETERINARY MEDICINAL PRODUCTS CETRIMIDE SUMMARY REPORT 1. Cetrimide (CAS 8044-71-1) is a quaternary ammonium antiseptic and surfactant. It consists mostly of trimethyltetradecylammonium bromide (CAS 1119-97-7; also known as tetradonium bromide) with smaller amounts of dodecyltrimethylammonium bromide (CAS 1119-94-4; also known as DTAB) and hexadecyltrimethylammonium bromide (CAS 57-09-0). The European Pharmacopoeia requires that cetrimide should contain at least 96% but no more than 101.0% alkyltrimethylammonium bromides. 2. Historically, the name cetrimide was used for a material consisting of predominantly hexadecyltrimethylammonium bromide, (CAS 57-09-0; also known as cetrimonium bromide, CTAB, and cetyltrimethylammonium bromide), together with smaller amounts of analogous alkyltrimethylammonium bromides. Some of the safety studies were carried out using this "cetrimide" rather than the cetrimide currently specified in the Europeam Pharmacopoeia. 3. In veterinary medicine, cetrimide is used as a topical antiseptic at concentrations of up to 2%. It is also used as an excipient in an injectable antibiotic formulation intended for use in cattle, sheep and pigs. When used as excipient, the concentration of cetrimide in the formulation is around 0.25 mg/ml, resulting in a dose of approximately 0.01 mg/kg bw of cetrimide in the target species. 4. In aqueous solution, cetrimide dissociates to a biologically-active cation and an inactive anion. Cetrimide is inactive towards bacterial spores, it is effective against some viruses and has variable anti-fungal activity. The cation is also responsible for the surfactant activity. -

Legemiddelforbruket I Norge 2012–2016 Drug Consumption in Norway 2012–2016
2017:1 LEGEMIDDELSTATISTIKK EVT STIKKTITTEL INN HER Legemiddelforbruket i Norge 2012–2016 Drug Consumption in Norway 2012–2016 ISSN 1890-9647 Legemiddelforbruket i Norge 2012–2016 Drug Consumption in Norway 2012–2016 Solveig Sakshaug Hanne Strøm Christian Berg Hege Salvesen Blix Irene Litleskare Tove Granum Utgitt av Folkehelseinstituttet / Published by Norwegian Institute of Public Health Området for Psykisk og fysisk helse Avdeling for Legemiddelepidemiologi Mars 2017 Tittel/Title: Legemiddelstatistikk 2017:1 Legemiddelforbruket i Norge 2012-2016/Drug Consumption in Norway 2012-2016 Forfattere/Authors: Solveig Sakshaug (redaktør) Hanne Strøm Christian Berg Hege Salvesen Blix Irene Litleskare Tove Granum Bestilling/Order: Rapporten kan lastes ned som pdf på Folkehelseinstituttets nettsider: www.fhi.no/ The report is available as pdf format only and can be downloaded from the www.fhi.no Layout omslag: www.fetetyper.no Kontaktinformasjon/Contact information Folkehelseinstituttet/Norwegian Institute of Public Health P.O.Box 4404 Nydalen N-0403 Oslo Tel: +47 21 07 70 00 ISSN: 1890-9647 ISBN 978-82-8082-826-2 elektronisk utgave Sitering/Citation: Sakshaug, S (red), Legemiddelforbruket i Norge 2012-2016 [Drug Consumption in Norway 2012-2016], Legemiddelstatistikk 2017:1, Oslo: Folkehelseinstituttet, 2017. Tidligere utgaver/Previous editions: 1977: Legemiddelforbruket i Norge 1974 - 1976 1978: Legemiddelforbruket i Norge 1975 - 1977 1980: Legemiddelforbruket i Norge 1975 - 1979 1981: Legemiddelforbruket i Norge 1980 1982: Legemiddelforbruket -

Povidone-Iodine (Topical)
PATIENT & CAREGIVER EDUCATION Povidone-Iodine (Topical) This information from Lexicomp® explains what you need to know about this medication, including what it’s used for, how to take it, its side effects, and when to call your healthcare provider. Brand Names: US Aplicare Povidone-Iodine Scrub [OTC] [DSC]; Aplicare Povidone-Iodine [OTC] [DSC]; Betadine Antiseptic Dry Powder [OTC]; Betadine Antiseptic Gargle [OTC]; Betadine Antiseptic Rinse [OTC]; Betadine Antiseptic [OTC]; Betadine Skin Cleanser [OTC] [DSC]; Betadine Surgical Scrub [OTC]; Betadine Swabsticks [OTC]; Betadine [OTC]; Clorox Nasal Antiseptic [OTC] [DSC]; ExCel AP [OTC] [DSC]; First Aid Antiseptic [OTC]; NuPrep 5% Povidone-Iodine [OTC] [DSC]; PVP Prep [OTC] [DSC]; PVP Scrub [OTC] [DSC]; Scrub Care Povidone-iodine [OTC]; Summers Eve Disp Medicated [OTC] What is this drug used for? All skin products: It is used to treat or prevent bacterial infections. It may be given to your child for other reasons. Talk with the doctor. All vaginal products: It is used to treat vaginal irritation, itching, and soreness. Throat gargle: It is used to treat a sore throat. Povidone-Iodine (Topical) 1/7 What do I need to tell the doctor BEFORE my child takes this drug? All products: If your child is allergic to this drug; any part of this drug; or any other drugs, foods, or substances. Tell the doctor about the allergy and what signs your child had. If your child is pregnant or breast-feeding a baby: If your child is pregnant or may be pregnant. Some forms of this drug are not for use during pregnancy. Talk with the doctor if your child is breast-feeding a baby or plans to breast- feed a baby. -

Antiseptics in the Era of Bacterial Resistance: a Focus on Povidone Iodine
Therapeutic Perspective Antiseptics in the era of bacterial resistance: a focus on povidone iodine Jean-Marie Lachapelle*1, Olivier Castel2, Alejandro Fueyo Casado3, Bernard Leroy1, Giuseppe Micali4, Dominique Tennstedt1 & Julien Lambert5 Practice Points Increasing bacterial resistance to antibiotics makes the management of superficial skin infections a major medical challenge. Antiseptics have broader spectrums of antimicrobial activity and a reduced potential for selection of bacterial resistance, relative to antibiotics. Consequently, antiseptics are appropriate alternatives to antibiotics for the prevention and treatment of superficial skin infections. Of four widely used antiseptics (povidone iodine, polihexanide, chlorhexidine and octenidine), povidone iodine has a particularly broad spectrum of antimicrobial activity that includes Gram-positive and Gram-negative bacteria, bacterial spores, fungi, protozoa and viruses. Widespread and extended use of povidone iodine is not associated with the selection of resistant bacterial strains. In contrast, bacterial resistance to chlorhexidine, quaternary ammonium salts, silver and triclosan has been documented. Regarding duration of effect on healthy skin, chlorhexidine is active for 1–4 h, whereas solutions of povidone iodine are active for 12–14 h. Aqueous and hydroalcoholic formulations of povidone iodine have good skin tolerance. Povidone iodine scrub has better skin tolerance than soap formulations of chlorhexidine and quaternary ammonium compounds (e.g., benzalkonium chloride and -

Intolerance of Chlorhexidine As a Skin Antiseptic in Patients Undergoing Hemodialysis Author(S): Alexander J
Intolerance of Chlorhexidine as a Skin Antiseptic in Patients Undergoing Hemodialysis Author(s): Alexander J. Kallen, Priti R. Patel, Sally Hess Source: Infection Control and Hospital Epidemiology, Vol. 32, No. 11 (November 2011), pp. 1144-1146 Published by: The University of Chicago Press on behalf of The Society for Healthcare Epidemiology of America Stable URL: http://www.jstor.org/stable/10.1086/662591 Accessed: 26/10/2011 14:03 Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at http://www.jstor.org/page/info/about/policies/terms.jsp JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range of content in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new forms of scholarship. For more information about JSTOR, please contact [email protected]. The University of Chicago Press and The Society for Healthcare Epidemiology of America are collaborating with JSTOR to digitize, preserve and extend access to Infection Control and Hospital Epidemiology. http://www.jstor.org 114 4 in fe ct io n co n tr ol and hos pi t al epi d em io lo g y nov embe r 20 11 , vol . 32, no. 11 Reporting Program. CDC will continue to provide tools for of Health Web site. http://www.health.ny.gov/statistics/facilities/ these patient safety efforts, and NHSN will evolve to help hospital/hospital_acquired_infections/2007/docs/hospital reduce the burden of data collection and inconsistencies be acquired_infection-full_report.pdf. -

Benefits of Benzalkonium Chloride Based Hand Sanitizers
Benzalkonium chloride-based Hand Sanitizers have distinct advantages over gelled alcohol hand sanitizers. While both product forms are FDA Monograph compliant for leave on products, fast acting and allow for use without water or towels, benzalkonium chloride-based products are non-flammable, less drying to skin, and will not stain clothing. Published studies report that benzalkonium chloride based hand sanitizers demonstrated greater sustained degerming activity than gelled alcohol gel hand sanitizers that actually became less effective with repeated use and made the skin dirtier, not cleaner due to removal of protective natural skin oils and entrapment of dead skin cells by the polymer thickeners used in the gelled alcohol products (AORN Journal, (68 August 1998), p. 239-251). Benzalkonium chloride, unlike benzethonium chloride, is the only quat active ingredient with a history of use in leave-on, FDA Monograph anti-bacterial skin treatment products. Leave-on Hand Sanitizers should not be used as a substitute for proper hand washing and hygiene practices. Patented Non-Alcohol Instant Foaming Hand Sanitizer produces a fast drying, non-sticky foam that contains unique non-drying, conditioning and moisturizing ingredients, leaves the skin with a soft, refreshing and silky after feel, and does not contain polymer thickeners or silicones. Multiple Instant Foam Hand Sanitizer, based on the active ingredient Benzalkonium chloride, featuring exceptional skin feel, conditioning and moisturizing properties. The efficacy of these product have been confirmed to reduce S. aureus 99.9999% in as little as 15 seconds. All of the Instant Foam Hand Sanitizer are in compliance with the FDA Final Tentative Monograph for OTC Hand Sanitizer preparations (leave-on sanitizers not requiring a rinse), and most are registered in Canada. -
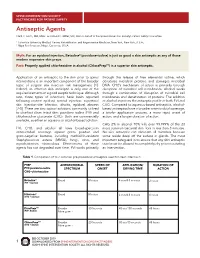
Antiseptic Agents Clark C
SPINE INTERVENTION SOCIETY FACTFINDERS FOR PATIENT SAFETY Antiseptic Agents Clark C. Smith, MD, MPH1 and David C. Miller, MD, MA2 on behalf of the Spine Intervention Society’s Patient Safety Committee 1 Columbia University Medical Center, Rehabilitation and Regenerative Medicine, New York, New York, U.S.A.; 2 Napa Pain Institute, Napa, California, U.S.A. Myth: For an epidural injection, Betadine® (povidone-iodine) is just as good a skin antiseptic as any of those modern expensive skin preps. Fact: Properly applied chlorhexidine in alcohol (ChloraPrepTM) is a superior skin antiseptic. Application of an antiseptic to the skin prior to spinal through the release of free elemental iodine, which interventions is an important component of the broader denatures microbial proteins and damages microbial topic of surgical site infection risk management [1]. DNA. CHG’s mechanism of action is primarily through Indeed, an effective skin antiseptic is only one of the disruption of microbial cell membranes. Alcohol works required elements of a good aseptic technique. Although through a combination of disruption of microbial cell rare, three types of infections have been reported membranes and denaturation of proteins. The addition following routine epidural steroid injection: superficial of alcohol improves the antiseptic profile of both PVI and skin injection-site infection, discitis, epidural abscess CHG. Compared to aqueous-based antiseptics, alcohol- [2-5]. There are two topical solutions commonly utilized based antiseptics have a broader antimicrobial coverage, to disinfect clean intact skin: povidone-iodine (PVI) and a briefer application process, a more rapid onset of chlorhexidine gluconate (CHG). Both are commercially action, and a longer duration of action. -

Medical Product Quality Report – COVID‐19 Issues Issue 2
Medical Product Quality Report – COVID‐19 Issues Issue 2. July 2020 The document has been produced by the Medicine Quality Research Group, Centre of Tropical Medicine & Global Health, Nuffield Department of Medicine, University of Oxford This report was prepared by Kerlijn Van Assche, Céline Caillet and Paul Newton of the Medicine Quality Research Group, that is part of the MORU Tropical Health Network and the Infectious Diseases Data Observatory (IDDO), Centre for Tropical Medicine & Global Health, Nuffield Department of Medicine, University of Oxford, UK. The Globe system was developed by Clark Freifeld (HealthMap, Boston Children’s Hospital, NorthEastern University) and Andrew Payne (IDDO), Alberto Olliaro (IDDO) and Gareth Blower (ex‐ IDDO) with curation of the English reports by Kem Boutsamay, Konnie Bellingham and Vayouly Vidhamaly, based in the Lao‐Oxford‐Mahosot Hospital‐Wellcome Trust Research Unit (LOMWRU), Microbiology Laboratory, Mahosot Hospital, Vientiane, Lao PDR. This document is open access but we would be grateful if you could cite it as: Medicine Quality Research Group, University of Oxford. Medical Product Quality Report – COVID‐19 Issues. Issue 2, July 2020. The work is kindly supported by the Bill&Melinda Gates Foundation, the University of Oxford and the Wellcome Trust. August 2020 2 Contents 1 Summary of findings ........................................................................................................................ 4 2 Introduction ....................................................................................................................................