The Complete Mitochondrial Genome of the Whiting, Merlangius
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Histological Aspects of the Early Development of the Digestive System of Burbot Lota Lota L
ISSN 0015-5497, e-ISSN 1734-9168 Folia Biologica (Kraków), vol. 64 (2016), No 1 Ó Institute of Systematics and Evolution of Animals, PAS, Kraków, 2015 doi:10.3409/fb64_1.11 Histological Aspects of the Early Development of the Digestive System of Burbot Lota lota L. (Lotidae, Gadiformes)* Gra¿yna FURGA£A-SELEZNIOW, Ma³gorzata JANKUN, Roman KUJAWA, Joanna NOWOSAD, Maria BI£AS, Dariusz KUCHARCZYK, and Andrzej SKRZYPCZAK Accepted December 04, 2015 Published January 29, 2016 FURGA£A-SELEZNIOW G., JANKUN M., KUJAWA R., NOWOSAD J., BI£AS M., KUCHARCZYK D., SKRZYPCZAK A. 2016. Histological aspects of the early development of the digestive system of burbot Lota lota L. (Lotidae, Gadiformes). Folia Biologica (Kraków) 64: 11-21. The ontogeny of the digestive tract was studied histologically in burbot, Lota lota L., from hatching to 42 days post-hatch (dph). At hatching, the digestive tract consisted of a straight tube with discernible digestive accessory glands (the liver and the pancreas) dorsally attached to the yolk sac. Most of the yolk sac reserves were consumed during the first 12 days and were completely depleted by 17 dph. The first PAS-positive goblet cells appeared at 6 dph, dispersed within the epithelium of the oesophagus and increasing substantially in number and distribution as development progressed. At 12 dph, the first vacuoles (neutral lipids) appeared in the intestine, indicating the functional absorption of nutrients from food. Differentiation of gastric glands was first noticed at 17 dph and was extensive by 27 dph. L. lota larvae have a morphologically complete digestive tract by 32 dph. -

Marine Fishes from Galicia (NW Spain): an Updated Checklist
1 2 Marine fishes from Galicia (NW Spain): an updated checklist 3 4 5 RAFAEL BAÑON1, DAVID VILLEGAS-RÍOS2, ALBERTO SERRANO3, 6 GONZALO MUCIENTES2,4 & JUAN CARLOS ARRONTE3 7 8 9 10 1 Servizo de Planificación, Dirección Xeral de Recursos Mariños, Consellería de Pesca 11 e Asuntos Marítimos, Rúa do Valiño 63-65, 15703 Santiago de Compostela, Spain. E- 12 mail: [email protected] 13 2 CSIC. Instituto de Investigaciones Marinas. Eduardo Cabello 6, 36208 Vigo 14 (Pontevedra), Spain. E-mail: [email protected] (D. V-R); [email protected] 15 (G.M.). 16 3 Instituto Español de Oceanografía, C.O. de Santander, Santander, Spain. E-mail: 17 [email protected] (A.S); [email protected] (J.-C. A). 18 4Centro Tecnológico del Mar, CETMAR. Eduardo Cabello s.n., 36208. Vigo 19 (Pontevedra), Spain. 20 21 Abstract 22 23 An annotated checklist of the marine fishes from Galician waters is presented. The list 24 is based on historical literature records and new revisions. The ichthyofauna list is 25 composed by 397 species very diversified in 2 superclass, 3 class, 35 orders, 139 1 1 families and 288 genus. The order Perciformes is the most diverse one with 37 families, 2 91 genus and 135 species. Gobiidae (19 species) and Sparidae (19 species) are the 3 richest families. Biogeographically, the Lusitanian group includes 203 species (51.1%), 4 followed by 149 species of the Atlantic (37.5%), then 28 of the Boreal (7.1%), and 17 5 of the African (4.3%) groups. We have recognized 41 new records, and 3 other records 6 have been identified as doubtful. -
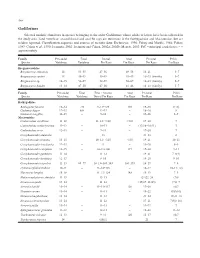
Gadiformes Selected Meristic Characters in Species Belonging to the Order Gadiformes Whose Adults Or Larvae Have Been Collected in the Study Area
548 Gadiformes Selected meristic characters in species belonging to the order Gadiformes whose adults or larvae have been collected in the study area. Total vertebrae, second dorsal and anal fin rays are numerous in the Bathygadidae and Macrouridae, but are seldom reported. Classification sequence and sources of meristic data: Eschmeyer, 1990; Fahay and Markle, 1984; Fahay, 1989; Cohen et al., 1990; Iwamoto, 2002; Iwamoto and Cohen, 2002a; 2002b; Merrett, 2003. PrC = principal caudal rays; ~ = approximately Family Precaudal Total Dorsal Anal Pectoral Pelvic Species Vertebrae Vertebrae Fin Rays Fin Rays Fin Rays Fin Rays Bregmacerotidae Bregmaceros atlanticus 14 53–55 47–56 49–58 16–21 5–7 Bregmaceros cantori 14 45–49 45–49 45–49 16–23 (family) 5–7 Bregmaceros sp. 14–15 52–59 52–59 58–69 16–23 (family) 5–7 Bregmaceros houdei 13–14 47–50 47–50 41–46 16–23 (family) 5–7 Family Precaudal Total First + Second Anal Pectoral Pelvic Species Vertebrae Vertebrae Dorsal Fin Rays Fin Rays Fin Rays Fin Rays Bathygadidae Bathygadus favosus 12–14 ~70 9–11+125 110 15–18 9(10) Gadomus dispar 12–13 80+ 12–13 – 18–20 8 Gadomus longifilis 11–13 – 9–11 – 14–16 8–9 Macrouridae Caelorinchus caribbeus 11–12 – 11–12+>110 >110 17–20 7 Caelorinchus coelorhynchus 11–12 – 10–11 – (17)18–20(21) 7 Caelorinchus occa 12–13 – 9–11 – 17–20 7 Coryphaenoides alateralis – 13 – 21–23 8 Coryphaenoides armatus 13–15 – 10–12+~125 ~135 19–21 10–11 Coryphaenoides brevibarbis 12–13 – 9 – 19–20 8–9 Coryphaenoides carapinus 12–15 – 10–11+100 117 17–20 9–11 Coryphaenoides guentheri -

Length-Weight Relationships of Marine Fish Collected from Around the British Isles
Science Series Technical Report no. 150 Length-weight relationships of marine fish collected from around the British Isles J. F. Silva, J. R. Ellis and R. A. Ayers Science Series Technical Report no. 150 Length-weight relationships of marine fish collected from around the British Isles J. F. Silva, J. R. Ellis and R. A. Ayers This report should be cited as: Silva J. F., Ellis J. R. and Ayers R. A. 2013. Length-weight relationships of marine fish collected from around the British Isles. Sci. Ser. Tech. Rep., Cefas Lowestoft, 150: 109 pp. Additional copies can be obtained from Cefas by e-mailing a request to [email protected] or downloading from the Cefas website www.cefas.defra.gov.uk. © Crown copyright, 2013 This publication (excluding the logos) may be re-used free of charge in any format or medium for research for non-commercial purposes, private study or for internal circulation within an organisation. This is subject to it being re-used accurately and not used in a misleading context. The material must be acknowledged as Crown copyright and the title of the publication specified. This publication is also available at www.cefas.defra.gov.uk For any other use of this material please apply for a Click-Use Licence for core material at www.hmso.gov.uk/copyright/licences/ core/core_licence.htm, or by writing to: HMSO’s Licensing Division St Clements House 2-16 Colegate Norwich NR3 1BQ Fax: 01603 723000 E-mail: [email protected] 3 Contents Contents 1. Introduction 5 2. -

Establishment and Partial Characterization of a Cell Line from Burbot Lota Lota Maculosa: Susceptibility to IHNV, IPNV and VHSV Mark P
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln USGS Staff -- ubP lished Research US Geological Survey 5-18-2010 Establishment and partial characterization of a cell line from burbot Lota lota maculosa: susceptibility to IHNV, IPNV and VHSV Mark P. Polinski Department of Fish and Wildlife Resources, College of Natural Resources, University of Idaho John D. Drennan Intervet/Schering-Plough Animal Health William N. Batts US Geological Survey, Western Fisheries Research Center Susan C. Ireland Kootenai Tribe of Idaho Kenneth D. Cain Department of Fish and Wildlife Resources, College of Natural Resources, University of Idaho,, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/usgsstaffpub Part of the Geology Commons, Oceanography and Atmospheric Sciences and Meteorology Commons, Other Earth Sciences Commons, and the Other Environmental Sciences Commons Polinski, Mark P.; Drennan, John D.; Batts, iW lliam N.; Ireland, Susan C.; and Cain, Kenneth D., "Establishment and partial characterization of a cell line from burbot Lota lota maculosa: susceptibility to IHNV, IPNV and VHSV" (2010). USGS Staff -- Published Research. 1062. http://digitalcommons.unl.edu/usgsstaffpub/1062 This Article is brought to you for free and open access by the US Geological Survey at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in USGS Staff -- ubP lished Research by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Vol. 90: 15–23, 2010 DISEASES OF AQUATIC ORGANISMS Published May 18 doi: 10.3354/dao02215 Dis Aquat Org Establishment and partial characterization of a cell line from burbot Lota lota maculosa: susceptibility to IHNV, IPNV and VHSV Mark P. -

Comparison of Size Selectivity Between Marine Mammals and Commercial Fisheries with Recommendations for Restructuring Management Policies
NOAA Technical Memorandum NMFS-AFSC-159 Comparison of Size Selectivity Between Marine Mammals and Commercial Fisheries with Recommendations for Restructuring Management Policies by M. A. Etnier and C. W. Fowler U.S. DEPARTMENT OF COMMERCE National Oceanic and Atmospheric Administration National Marine Fisheries Service Alaska Fisheries Science Center October 2005 NOAA Technical Memorandum NMFS The National Marine Fisheries Service's Alaska Fisheries Science Center uses the NOAA Technical Memorandum series to issue informal scientific and technical publications when complete formal review and editorial processing are not appropriate or feasible. Documents within this series reflect sound professional work and may be referenced in the formal scientific and technical literature. The NMFS-AFSC Technical Memorandum series of the Alaska Fisheries Science Center continues the NMFS-F/NWC series established in 1970 by the Northwest Fisheries Center. The NMFS-NWFSC series is currently used by the Northwest Fisheries Science Center. This document should be cited as follows: Etnier, M. A., and C. W. Fowler. 2005. Comparison of size selectivity between marine mammals and commercial fisheries with recommendations for restructuring management policies. U.S. Dep. Commer., NOAA Tech. Memo. NMFS-AFSC-159, 274 p. Reference in this document to trade names does not imply endorsement by the National Marine Fisheries Service, NOAA. NOAA Technical Memorandum NMFS-AFSC-159 Comparison of Size Selectivity Between Marine Mammals and Commercial Fisheries with Recommendations for Restructuring Management Policies by M. A. Etnier and C. W. Fowler Alaska Fisheries Science Center 7600 Sand Point Way N.E. Seattle, WA 98115 www.afsc.noaa.gov U.S. DEPARTMENT OF COMMERCE Carlos M. -

HANDBOOK of FISH BIOLOGY and FISHERIES Volume 1 Also Available from Blackwell Publishing: Handbook of Fish Biology and Fisheries Edited by Paul J.B
HANDBOOK OF FISH BIOLOGY AND FISHERIES Volume 1 Also available from Blackwell Publishing: Handbook of Fish Biology and Fisheries Edited by Paul J.B. Hart and John D. Reynolds Volume 2 Fisheries Handbook of Fish Biology and Fisheries VOLUME 1 FISH BIOLOGY EDITED BY Paul J.B. Hart Department of Biology University of Leicester AND John D. Reynolds School of Biological Sciences University of East Anglia © 2002 by Blackwell Science Ltd a Blackwell Publishing company Chapter 8 © British Crown copyright, 1999 BLACKWELL PUBLISHING 350 Main Street, Malden, MA 02148‐5020, USA 108 Cowley Road, Oxford OX4 1JF, UK 550 Swanston Street, Carlton, Victoria 3053, Australia The right of Paul J.B. Hart and John D. Reynolds to be identified as the Authors of the Editorial Material in this Work has been asserted in accordance with the UK Copyright, Designs, and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs, and Patents Act 1988, without the prior permission of the publisher. First published 2002 Reprinted 2004 Library of Congress Cataloging‐in‐Publication Data has been applied for. Volume 1 ISBN 0‐632‐05412‐3 (hbk) Volume 2 ISBN 0‐632‐06482‐X (hbk) 2‐volume set ISBN 0‐632‐06483‐8 A catalogue record for this title is available from the British Library. Set in 9/11.5 pt Trump Mediaeval by SNP Best‐set Typesetter Ltd, Hong Kong Printed and bound in the United Kingdom by TJ International Ltd, Padstow, Cornwall. -

The Genetic Diversity of Burbot (Lota Lota L., 1758) of Western Siberia (The Analysis of the Mtdna Control Region Polymorphism)1 Yu
ISSN 1022-7954, Russian Journal of Genetics, 2017, Vol. 53, No. 2, pp. 233–241. © Pleiades Publishing, Inc., 2017. Published in Russian in Genetika, 2017, Vol. 53, No. 2, pp. 214–222. ANIMAL GENETICS The Genetic Diversity of Burbot (Lota lota L., 1758) of Western Siberia (the Analysis of the mtDNA Control Region Polymorphism)1 Yu. Ya. Khrunyk, V. D. Bogdanov, L. E. Yalkovskaya, A. R. Koporikov, S. B. Rakitin, P. A. Sibiryakov, and A. V. Borodin* Institute of Plant and Animal Ecology, Ural Branch, Russian Academy of Sciences, Yekaterinburg, 620144 Russia *e-mail: [email protected] Received March 3, 2016; in final form, April 19, 2016 AbstractThe genetic variability of burbot (Lota lota L., 1758) inhabiting the Ob-Irtysh and Taz river basins in Western Siberia has been studied based on the polymorphism of the hypervariable fragment of mtDNA control region (407 bp). The analysis of 134 fish samples revealed 30 haplotypes, 23 of which were new. Among haplotypes, previously detected in Eurasia and North America, EB30 was the most frequently found in Western Siberia (45.5% frequency). The results of our study are in agreement with previous research point- ing to the genetic differentiation of two burbot subspecies, L. l. lota and L. l. maculosa, and indicate that bur- bot inhabiting the Ob-Irtysh and Taz river basins belong to the Eurasian-Beringian clade (nominative sub- species L. l. lota). However, a high genetic diversity of burbot in Western Siberia, along with a relatively high differentiation of burbot groups within studied territory, points to a regional specificity of burbot population. -

A Recovery/Conservation Programme for Marine Species of Conservation Importance
Natural England Commissioned Report NECR065 A recovery/conservation programme for marine species of conservation importance First published 20 December 2011 www.naturalengland.org.uk Foreword Natural England commission a range of reports from external contractors to provide evidence and advice to assist us in delivering our duties. The views in this report are those of the authors and do not necessarily represent those of Natural England. Background Natural England commissioned this project to should result in the relevant species becoming provide an auditable, transparent methodology self-sustaining members of their ecosystems. for prioritising marine species recovery programmes in the UK waters around England. This report should be cited as: The aim of the work is to identify the HISCOCK, K., BAYLEY, D., PADE, N., COX, E. conservation needs of all marine Biodiversity & LACEY, C. 2011. A recovery / conservation Action Plan (BAP), OSPAR and WCA species in programme for marine species of conservation English waters, that are not considered to be importance. Natural England Commissioned afforded sufficient protection by the forthcoming Reports, Number 065. Marine Protected Areas network. The results will be used by Natural England to improve our understanding the marine species that are most in need of targeted conservation action. The report focuses on designing a prioritisation methodology that will be used to inform evidence based and transparent decision making regarding species recovery in the marine environment and where possible, -

Studies on the Biology of Non-Commercial Species, Based on The
10.1515/umcsbio-2015-0008 A N N A L E S U N I V E R S I T A T I S M A R I A E C U R I E - S K Ł O D O W S K A L U B L I N – P O L O N I A VOL. LXX, 1 SECTIO C 2015 MAGDALENA Lampart- KAŁUżNIACKA, TOMASz HEESE University of Technology, Division of Environmental Biology 75-453 Koszalin, ul. śniadeckich 2; e-mail: [email protected] Studies on the biology of non-commercial species, based on the example of the fourbeard rockling Enchelyopus cimbrius (L., 1766) (Gadiformes: Lotidae) in the southern Baltic Badania biologiczne niekomercyjnego gatunku, na przykładzie moteli Fourbeard Rockling Enchelyopus cimbrius (L., 1766) (Gadiformes: Lotidae) w południowym Bałtyku. ABSTRACT The paper includes a detailed analysis of selected elements of the biology of the forebeard rockling. The study was conducted in the southern Baltic Sea. The sex structure, fertility, stomach contents, age, and growth of the species were analysed. As a result of the study, double predominance in numbers of males over females and high diversity in terms of development and size of oocytes were determined, suggesting partial spawning. A strong correlation was recorded between fertility and body mass and length. The analysis of the content of the fish stomachs revealed dominance of Polychaeta, Crustacea, and fish. Analyses of age and growth were also conducted, based on the otoliths. Individuals of 4 to 13 years of age were found. In the samples analysed, males were older than females. -

HELCOM Red List of Threatened and Declining Species of Lampreys and Fishes of the Baltic Sea
Baltic Sea Environment Proceedings No. 109 HELCOM Red list of threatened and declining species of lampreys and fishes of the Baltic Sea Helsinki Commission Baltic Marine Environment Protection Commission Baltic Sea Environment Proceedings No. 109 HELCOM Red list of threatened and declining species of lampreys and fishes of the Baltic Sea Helsinki Commission Baltic Marine Environment Protection Commission Editor: Dr. Ronald Fricke, Curator of fishes, Ichtyology Contact address: Staatliches Museum für Naturkunde Stuttgart Rosenstein 1, 70191 Stuttgart, Germany E-mail: [email protected] Photographs © BfN, Krause & Hübner. Cover photo: Gobius niger For bibliographic purposes this document should be cited to as: HELCOM 2007: HELCOM Red list of threatened and declining species of lampreys and fish of the Baltic Sea. Baltic Sea Environmental Proceedings, No. 109, 40 pp. Information included in this publication or extracts there of is free for citing on the condition that the complete reference of the publication is given as stated above. Copyright 2007 by the Baltic Marine Environment Protection Commission - Helsinki Commission ISSN 0357-2944 Table of Contents 1 Introduction .......................................................................................................................6 2 Species and area covered.............................................................................................7 2.1 Species covered..............................................................................................................7 -

Early Opening Experimental Fishery for Silver Hake/ Whiting in Small Mesh Area 1 and the Western Raised Footrope Exemption Area
Massachusetts Division of Marine Fisheries Early Opening Experimental Fishery for Silver Hake/ Whiting in Small Mesh Area 1 and the Western Raised Footrope Exemption Area David M. Chosid1, Michael Pol1, Bradley P. Schondelmeier2, and Melanie Griffin3 Massachusetts Division of Marine Fisheries 1836 S. Rodney French Blvd. New Bedford, MA 02774 230 Emerson Ave. Gloucester, MA 01930 3251 Causeway St., Suite 400 Boston, MA 02114 July 2018 Commonwealth of Massachusetts Charles D. Baker, Governor Executive Office of Energy and Environmental Affairs Matthew A. Beaton, Secretary Department of Fish and Game Ronald S. Amidon, Commissioner Massachusetts Division of Marine Fisheries David E. Pierce, PhD, Director Executive Summary It may be possible to reduce bycatch by working with fishermen. Some vessels caught either more or less by- Times and areas where small mesh codends can be catch, which could be due to small differences in the used to fish for whiting (silver hake) have not changed RFT, in tow location, or other factors. The date, time of since the 1990s although fish migrations have shifted. day, or the order of the tow did not affect discards. Fur- Fishermen asked the Massachusetts Division of Marine ther, new gear modifications could be developed and Fisheries (DMF) to help test early openings of Small tested. Mesh Area 1 (SMA1) in Ipswich Bay and the Western Raised Footrope Exemption (WRFE) area in Cape Cod Fishermen are still interested in an early opening of Bay because they observed whiting appearing earlier SMA1. The data collected during the experimental fish- than before. Using funds from the Groundfish Disas- ery can help weigh the fishing benefits and bycatch ter Aid Program, DMF organized experimental fisheries risks.