Determination of Thiazinamium, Promazine and Promethazine in Pharmaceutical Formulations Using a CZE Method Francisco J
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Drug Use Evaluation: Antipsychotic Utilization in Schizophrenia Patients
© Copyright 2012 Oregon State University. All Rights Reserved Drug Use Research & Management Program Oregon State University, 500 Summer Street NE, E35 Salem, Oregon 97301-1079 Phone 503-947-5220 | Fax 503-947-1119 Drug Use Evaluation: Antipsychotic Utilization in Schizophrenia Patients Research Questions: 1. How many schizophrenia patients are prescribed recommended first-line second-generation treatments for schizophrenia? 2. How many schizophrenia patients switch to an injectable antipsychotic after stabilization on an oral antipsychotic? 3. How many schizophrenia patients are prescribed 2 or more concomitant antipsychotics? 4. Are claims for long-acting injectable antipsychotics primarily billed as pharmacy or physician administered claims? 5. Does adherence to antipsychotic therapy differ between patients with claims for different routes of administration (oral vs. long-acting injectable)? Conclusions: In total, 4663 schizophrenia patients met inclusion criteria, and approximately 14% of patients (n=685) were identified as treatment naïve without claims for antipsychotics in the year before their first antipsychotic prescription. Approximately 45% of patients identified as treatment naïve had a history of remote antipsychotic use, but it is unclear if antipsychotics were historically prescribed for schizophrenia. Oral second-generation antipsychotics which are recommended as first-line treatment in the MHCAG schizophrenia algorithm were prescribed as initial treatment in 37% of treatment naive patients and 28% of all schizophrenia patients. Recommended agents include risperidone, paliperidone, and aripiprazole. Utilization of parenteral antipsychotics was limited in patients with schizophrenia. Overall only 8% of patients switched from an oral to an injectable therapy within 6 months of their first claim. Approximately, 60% of all schizophrenia patients (n=2512) had claims for a single antipsychotic for at least 12 continuous weeks and may be eligible to transition to a long-acting injectable antipsychotic. -

Schizophrenia Care Guide
August 2015 CCHCS/DHCS Care Guide: Schizophrenia SUMMARY DECISION SUPPORT PATIENT EDUCATION/SELF MANAGEMENT GOALS ALERTS Minimize frequency and severity of psychotic episodes Suicidal ideation or gestures Encourage medication adherence Abnormal movements Manage medication side effects Delusions Monitor as clinically appropriate Neuroleptic Malignant Syndrome Danger to self or others DIAGNOSTIC CRITERIA/EVALUATION (PER DSM V) 1. Rule out delirium or other medical illnesses mimicking schizophrenia (see page 5), medications or drugs of abuse causing psychosis (see page 6), other mental illness causes of psychosis, e.g., Bipolar Mania or Depression, Major Depression, PTSD, borderline personality disorder (see page 4). Ideas in patients (even odd ideas) that we disagree with can be learned and are therefore not necessarily signs of schizophrenia. Schizophrenia is a world-wide phenomenon that can occur in cultures with widely differing ideas. 2. Diagnosis is made based on the following: (Criteria A and B must be met) A. Two of the following symptoms/signs must be present over much of at least one month (unless treated), with a significant impact on social or occupational functioning, over at least a 6-month period of time: Delusions, Hallucinations, Disorganized Speech, Negative symptoms (social withdrawal, poverty of thought, etc.), severely disorganized or catatonic behavior. B. At least one of the symptoms/signs should be Delusions, Hallucinations, or Disorganized Speech. TREATMENT OPTIONS MEDICATIONS Informed consent for psychotropic -

Thioridazine Induces Apoptosis of Multidrug-Resistant Mouse Lymphoma Cells Transfected with the Human ABCB1 and Inhibits the Expression of P-Glycoprotein
ANTICANCER RESEARCH 31: 4201-4206 (2011) Thioridazine Induces Apoptosis of Multidrug-resistant Mouse Lymphoma Cells Transfected with the Human ABCB1 and Inhibits the Expression of P-Glycoprotein GABRIELLA SPENGLER1,3, JOSEPH MOLNAR1, MIGUEL VIVEIROS2,4 and LEONARD AMARAL2,3,4 1Institute of Medical Microbiology and Immunobiology, Faculty of Medicine, University of Szeged, Szeged, Hungary; 2Group of Mycobacteriology, Unit of Microbiology and 3Unit of Parasitology and Medical Microbiology (UPMM), Institute of Hygiene and Tropical Medicine, Universidade Nova de Lisboa, Lisboa, Portugal; 4Cost Action BM0701 (ATENS) of the European Commission, Brussels, Belgium Abstract. Aim: Chlorpromazine has activity against a large therapeutic aspects, and low availability of effective but also variety of cancer types. However, this phenothiazine produces costly forms of therapy (chemotherapy, radiotherapy). a plethora of serious side-effects. We have studied thioridazine Regardless of economic support, chemotherapy of cancer is (TZ), a phenothiazine neuroleptic that is much milder, for highly problematic, especially when therapy promotes the activity against multidrug-resistant (MDR) cancer cells, as development of multidrug resistance (MDR), hence cancer well as against the overexpressed ABCB1 transporter (P- becomes refractory not only to the initial chemotherapeutic glycoprotein) that is the cause for the MDR phenotype of these agent, but to many other anticancer drugs as well (1, 2). The cancer cells. Materials and Methods: MDR mouse T- development of an MDR phenotype of cancer cell has been lymphoma cells, transfected with the human gene ABCB1 that known for many decades to be due to the overexpression of codes for the transporter ABCB1, were incubated with TZ for transporters that extrude the anticancer agent before it various periods of time and examined for evidence of reaches its intended target (3). -

FDA Approved Drugs with Broad Anti-Coronaviral Activity Inhibit SARS-Cov-2 in Vitro
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.25.008482; this version posted March 27, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. FDA approved drugs with broad anti-coronaviral activity inhibit SARS-CoV-2 in vitro Stuart Weston1, Rob Haupt1, James Logue1, Krystal Matthews1 and Matthew B. Frieman*1 1 - Department of Microbiology and Immunology, University of Maryland School of Medicine, 685 W. Baltimore St., Room 380, Baltimore, MD, 21201, USA *Corresponding author. Email: [email protected] Key words: SARS-CoV-2, nCoV-2019, COVID-19, drug repurposing, FDA approved drugs, antiviral therapeutics, pandemic, chloroquine, hydroxychloroquine AbstraCt SARS-CoV-2 emerged in China at the end of 2019 and has rapidly become a pandemic with over 400,000 recorded COVID-19 cases and greater than 19,000 recorded deaths by March 24th, 2020 (www.WHO.org) (1). There are no FDA approved antivirals or vaccines for any coronavirus, including SARS-CoV-2 (2). Current treatments for COVID-19 are limited to supportive therapies and off-label use of FDA approved drugs (3). Rapid development and human testing of potential antivirals is greatly needed. A potentially quicker way to test compounds with antiviral activity is through drug re-purposing (2, 4). Numerous drugs are already approved for use in humans and subsequently there is a good understanding of their safety profiles and potential side effects, making them easier to test in COVID-19 patients. -

A Drug Repositioning Approach Identifies Tricyclic Antidepressants As Inhibitors of Small Cell Lung Cancer and Other Neuroendocrine Tumors
Published OnlineFirst September 26, 2013; DOI: 10.1158/2159-8290.CD-13-0183 RESEARCH ARTICLE A Drug Repositioning Approach Identifi es Tricyclic Antidepressants as Inhibitors of Small Cell Lung Cancer and Other Neuroendocrine Tumors Nadine S. Jahchan 1 , 2 , Joel T. Dudley 1 , Pawel K. Mazur 1 , 2 , Natasha Flores 1 , 2 , Dian Yang 1 , 2 , Alec Palmerton 1 , 2 , Anne-Flore Zmoos 1 , 2 , Dedeepya Vaka 1 , 2 , Kim Q.T. Tran 1 , 2 , Margaret Zhou 1 , 2 , Karolina Krasinska 3 , Jonathan W. Riess 4 , Joel W. Neal 5 , Purvesh Khatri 1 , 2 , Kwon S. Park 1 , 2 , Atul J. Butte 1 , 2 , and Julien Sage 1 , 2 Downloaded from cancerdiscovery.aacrjournals.org on September 29, 2021. © 2013 American Association for Cancer Research. Published OnlineFirst September 26, 2013; DOI: 10.1158/2159-8290.CD-13-0183 ABSTRACT Small cell lung cancer (SCLC) is an aggressive neuroendocrine subtype of lung cancer with high mortality. We used a systematic drug repositioning bioinformat- ics approach querying a large compendium of gene expression profi les to identify candidate U.S. Food and Drug Administration (FDA)–approved drugs to treat SCLC. We found that tricyclic antidepressants and related molecules potently induce apoptosis in both chemonaïve and chemoresistant SCLC cells in culture, in mouse and human SCLC tumors transplanted into immunocompromised mice, and in endog- enous tumors from a mouse model for human SCLC. The candidate drugs activate stress pathways and induce cell death in SCLC cells, at least in part by disrupting autocrine survival signals involving neurotransmitters and their G protein–coupled receptors. The candidate drugs inhibit the growth of other neuroendocrine tumors, including pancreatic neuroendocrine tumors and Merkel cell carcinoma. -

Light Scattering Studies of Amphiphilic Drugs Promethazine Hydrochloride and Imipramine Hydrochloride in Aqueous Electrolyte Solutions
12962 J. Phys. Chem. B 2008, 112, 12962–12967 Light Scattering Studies of Amphiphilic Drugs Promethazine Hydrochloride and Imipramine Hydrochloride in Aqueous Electrolyte Solutions Md. Sayem Alam,† Goutam Ghosh,‡ and Kabir-ud-Din*,† Department of Chemistry, Aligarh Muslim UniVersity, Aligarh-202 002, India, and UGC-DAE Consortium for Scientific Research, Trombay, Mumbai- 400 085, India ReceiVed: May 13, 2008; ReVised Manuscript ReceiVed: August 1, 2008 Two amphiphilic drugs, promethazine hydrochloride (PMT) and imipramine hydrochloride (IMP), have been studied using both static and dynamic light scattering techniques. Due to having rigid tricyclic hydrophobic moieties in their molecules, these drugs show interesting association behavior. The static light scattering (SLS) measurements show that the self-association commenced above a well-defined critical micellar concentration (cmc), which decreases with increasing NaBr concentration. The Gibbs energy of micellization, 0 ∆GM, in all cases, is negative. The colloidal stability of the system in terms of the interparticle interaction at different NaBr concentrations was studied using the dynamic light scattering (DLS) technique. The experimentally evaluated interparticle interaction parameter (kD) was compared with the Derjaguin-Landau- Verwey-Overbeek (DLVO) model. Interestingly, these two drugs with similar molecular structure show difference in their interparticle interactions, e.g., PMT showed complete agreement with the DLVO model whereas IMP showed clear deviation from this model at lower concentrations and agreement at higher concentrations of NaBr. 1. Introduction SCHEME 1: Molecular Structure of Promethazine Hydrochloride, PMT (a), and Imipramine Hydrochloride, Many drug molecules are amphiphilic and self-associate in IMP (b) a surfactant-like manner in aqueous environment to form small aggregates. -

HALDOL Decanoate 50 (Haloperidol)
HALDOL® Decanoate 50 (haloperidol) HALDOL® Decanoate 100 (haloperidol) For IM Injection Only WARNING Increased Mortality in Elderly Patients with Dementia-Related Psychosis Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Analyses of seventeen placebo-controlled trials (modal duration of 10 weeks), largely in patients taking atypical antipsychotic drugs, revealed a risk of death in drug-treated patients of between 1.6 to 1.7 times the risk of death in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in drug-treated patients was about 4.5%, compared to a rate of about 2.6% in the placebo group. Although the causes of death were varied, most of the deaths appeared to be either cardiovascular (e.g., heart failure, sudden death) or infectious (e.g., pneumonia) in nature. Observational studies suggest that, similar to atypical antipsychotic drugs, treatment with conventional antipsychotic drugs may increase mortality. The extent to which the findings of increased mortality in observational studies may be attributed to the antipsychotic drug as opposed to some characteristic(s) of the patients is not clear. HALDOL Decanoate is not approved for the treatment of patients with dementia-related psychosis (see WARNINGS). DESCRIPTION Haloperidol decanoate is the decanoate ester of the butyrophenone, HALDOL (haloperidol). It has a markedly extended duration of effect. It is available in sesame oil in sterile form for intramuscular (IM) injection. The structural formula of haloperidol decanoate, 4-(4-chlorophenyl)-1-[4-(4-fluorophenyl)-4-oxobutyl]-4 piperidinyl decanoate, is: Haloperidol decanoate is almost insoluble in water (0.01 mg/mL), but is soluble in most organic solvents. -
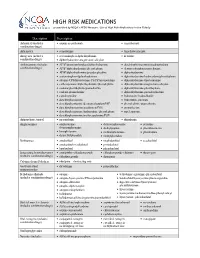
HIGH RISK MEDICATIONS As Specified by NCQA’S HEDIS Measure: Use of High Risk Medications in the Elderly
HIGH RISK MEDICATIONS as specified by NCQA’s HEDIS Measure: Use of High Risk Medications in the Elderly Description Prescription Antianxiety (includes • aspirin-meprobamate • meprobamate combination drugs) Antiemetics • scopolamine • trimethobenzamide Analgesics (includes • acetaminophen-diphenhydramine • ketorolac combination drugs) • diphenhydramine-magnesium salicylate Antihistamines (includes • APAP/dextromethorphan/diphenhydramine • dexchlorpheniramine-pseudoephedrine combination drugs) • APAP/diphenhydramine/phenylephrine • dextromethorphan-promethazine • APAP/diphenhydramine/pseudoephedrine • diphenhydramine • acetaminophen-diphenhydramine • diphenhydramine/hydrocodone/phenylephrine • atropine/CPM/hyoscyamine/PE/PPA/scopolamine • diphenhydramine-tripelennamine • carbetapentane/diphenhydramine/phenylephrine • diphenhydramine-magnesium salicylate • codeine/phenylephrine/promethazine • diphenhydramine-phenylephrine • codeine-promethazine • diphenhydramine-pseudoephedrine • cyproheptadine • hydroxyzine hydrochloride • dexchlorpheniramine • hydroxyzine pamoate • dexchlorpheniramine/dextromethorphan/PSE • phenylephrine-promethazine • dexchlorpheniramine/guaifenesin/PSE • promethazine • dexchlorpheniramine/hydrocodone/phenylephrine • tripelennamine • dexchlorpheniramine/methscopolamine/PSE Antipsychotic, typical • mesoridazine • thioridazine Amphetamines • amphetamine- • dextroamphetamine • pemoline dextroamphetamine • diethylpropion • phendimetrazine • benzphetamine • methamphetamine • phentermine • dexmethylphenidate • methylphenidate -

THIORIDAZINE HYDROCHLORIDE- Thioridazine Hydrochloride Tablet, Film Coated Mylan Institutional Inc
THIORIDAZINE HYDROCHLORIDE- thioridazine hydrochloride tablet, film coated Mylan Institutional Inc. ---------- WARNING Thioridazine has been shown to prolong the QTc interval in a dose related manner, and drugs with this potential, including thioridazine, have been associated with Torsades de pointes type arrhythmias and sudden death. Due to its potential for significant, possibly life threatening, proarrhythmic effects, thioridazine should be reserved for use in the treatment of schizophrenic patients who fail to show an acceptable response to adequate courses of treatment with other antipsychotic drugs, either because of insufficient effectiveness or the inability to achieve an effective dose due to intolerable adverse effects from those drugs (see WARNINGS, CONTRAINDICATIONS, and INDICATIONS). Increased Mortality in Elderly Patients with Dementia-Related Psychosis Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Analyses of seventeen placebo-controlled trials (modal duration of 10 weeks), largely in patients taking atypical antipsychotic drugs, revealed a risk of death in drug-treated patients of between 1.6 to 1.7 times the risk of death in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in drug- treated patients was about 4.5%, compared to a rate of about 2.6% in the placebo group. Although the causes of death were varied, most of the deaths appeared to be either cardiovascular (e.g., heart failure, sudden death) or infectious (e.g., pneumonia) in nature. Observational studies suggest that, similar to atypical antipsychotic drugs, treatment with conventional antipsychotic drugs may increase mortality. The extent to which the findings of increased mortality in observational studies may be attributed to the antipsychotic drug as opposed to some characteristic(s) of the patients is not clear. -

Neuromodulators for Functional Gastrointestinal Disorders (Disorders of Gutlbrain Interaction): a Rome Foundation Working Team Report Douglas A
Gastroenterology 2018;154:1140–1171 SPECIAL REPORT Neuromodulators for Functional Gastrointestinal Disorders (Disorders of GutLBrain Interaction): A Rome Foundation Working Team Report Douglas A. Drossman,1,2 Jan Tack,3 Alexander C. Ford,4,5 Eva Szigethy,6 Hans Törnblom,7 and Lukas Van Oudenhove8 1Center for Functional Gastrointestinal and Motility Disorders, University of North Carolina, Chapel Hill, North Carolina; 2Center for Education and Practice of Biopsychosocial Care and Drossman Gastroenterology, Chapel Hill, North Carolina; 3Translational Research Center for Gastrointestinal Disorders, University of Leuven, Leuven, Belgium; 4Leeds Institute of Biomedical and Clinical Sciences, University of Leeds, Leeds, United Kingdom; 5Leeds Gastroenterology Institute, St James’s University Hospital, Leeds, United Kingdom; 6Departments of Psychiatry and Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania; 7Departments of Internal Medicine and Clinical Nutrition, Institute of Medicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden; and 8Laboratory for BrainÀGut Axis Studies, Translational Research Center for Gastrointestinal Disorders, University of Leuven, Leuven, Belgium BACKGROUND & AIMS: Central neuromodulators (antide- summary information and guidelines for the use of central pressants, antipsychotics, and other central nervous neuromodulators in the treatment of chronic gastrointestinal systemÀtargeted medications) are increasingly used for treat- symptoms and FGIDs. Further studies are needed to confirm ment -

Promethazine-Chlorpromazine Combination in the Treatment of Unmanageable Psychotic Patients Armando R
Henry Ford Hospital Medical Journal Volume 17 | Number 4 Article 10 12-1969 Promethazine-Chlorpromazine Combination in the Treatment of Unmanageable Psychotic Patients Armando R. Favazza Follow this and additional works at: https://scholarlycommons.henryford.com/hfhmedjournal Part of the Chemicals and Drugs Commons, Life Sciences Commons, Medical Specialties Commons, Psychiatry and Psychology Commons, and the Public Health Commons Recommended Citation Favazza, Armando R. (1969) "Promethazine-Chlorpromazine Combination in the Treatment of Unmanageable Psychotic Patients," Henry Ford Hospital Medical Journal : Vol. 17 : No. 4 , 305-310. Available at: https://scholarlycommons.henryford.com/hfhmedjournal/vol17/iss4/10 This Article is brought to you for free and open access by Henry Ford Health System Scholarly Commons. It has been accepted for inclusion in Henry Ford Hospital Medical Journal by an authorized editor of Henry Ford Health System Scholarly Commons. Henry Ford Hosp. Med. Journal VoL 17, No. 4, 1969 Promethazine-chlorpromazine Combination in the Treatment of Unmanageable Psychotic Patients Armando R. Favazza, M.D.* Administering a combination of promethazine and chlorpromazine to patients with a "galloping psychosis" has an antipsychotic and tranquilizing effect which calms them down to a more manageable and less aggressive state. The drugs are chemically similar; promethazine's actions are strongly potentiated in the combina tion, so large doses must be given under careful supervision. Case histories demonstrate successful short term management of acutely psychotic, aggressive patients who were a danger to themselves and others. Acute, intense psychotic episodes common in the author's experience for can pose difficult problems for both some patients to receive massive doses the patient and those who care for him. -

Medications to Be Avoided Or Used with Caution in Parkinson's Disease
Medications To Be Avoided Or Used With Caution in Parkinson’s Disease This medication list is not intended to be complete and additional brand names may be found for each medication. Every patient is different and you may need to take one of these medications despite caution against it. Please discuss your particular situation with your physician and do not stop any medication that you are currently taking without first seeking advice from your physician. Most medications should be tapered off and not stopped suddenly. Although you may not be taking these medications at home, one of these medications may be introduced while hospitalized. If a hospitalization is planned, please have your neurologist contact your treating physician in the hospital to advise which medications should be avoided. Medications to be avoided or used with caution in combination with Selegiline HCL (Eldepryl®, Deprenyl®, Zelapar®), Rasagiline (Azilect®) and Safinamide (Xadago®) Medication Type Medication Name Brand Name Narcotics/Analgesics Meperidine Demerol® Tramadol Ultram® Methadone Dolophine® Propoxyphene Darvon® Antidepressants St. John’s Wort Several Brands Muscle Relaxants Cyclobenzaprine Flexeril® Cough Suppressants Dextromethorphan Robitussin® products, other brands — found as an ingredient in various cough and cold medications Decongestants/Stimulants Pseudoephedrine Sudafed® products, other Phenylephrine brands — found as an ingredient Ephedrine in various cold and allergy medications Other medications Linezolid (antibiotic) Zyvox® that inhibit Monoamine oxidase Phenelzine Nardil® Tranylcypromine Parnate® Isocarboxazid Marplan® Note: Additional medications are cautioned against in people taking Monoamine oxidase inhibitors (MAOI), including other opioids (beyond what is mentioned in the chart above), most classes of antidepressants and other stimulants (beyond what is mentioned in the chart above).