Product Retrieval Procedures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GLOBAL PRODUCT RECALL FOURTH EDITION Handbook
GLOBAL PRODUCT RECALL FOURTH EDITION Handbook Global Product Recall Handbook Fourth Edition Global Product Recall Handbook | Fourth Edition Foreword Baker McKenzie was founded in 1949. For almost seven decades, we have provided nuanced, sophisticated advice and leading-edge legal services to many of the world’s most dynamic and successful business organizations. With more than 7,000 internationally experienced lawyers in 47 countries, including 36 of the world’s largest economies, Baker McKenzie provides expertise in all of the substantive disciplines needed to formulate, develop and implement a global product recall. Our fluency in working across borders, issues and practices allows us to simplify legal complexity, foresee regulatory, legal, compliance, reputational, and commercial risks others may overlook and identify commercial opportunities that many miss. Taken together, this combination of deep practical experience and technical and substantive skills makes us advisers of choice to many of the world’s leading multinational corporations. Our clients want a new breed of lawyers with excellent technical skills and industry expertise who can look ahead to help them navigate a constantly changing product regulatory landscape. It means having lawyers who can anticipate what is coming next and provide practical legal resources that are helpful to the business at all levels. The Global Product Recall Handbook is one such resource, collecting, combining and synthesizing the advice of lawyers throughout Baker McKenzie focused on the consumer goods, pharmaceutical and medical devices, food and beverage, and motor vehicle industries. We are pleased also to make this edition of the Handbook available on a dedicated Dynamic Publisher site and accompanying mobile app. -

Nov. 2016 Edition OFFICE LOCATION
CONSUMER News Nov. 2016 Edition PROTECTION Dear Wisconsin Children’s Safety Advocates: In October 2016, the U.S. Consumer Product Safety Commission issued a total of 8 recalls relating to products affecting children. Attached is a summary of the releases identifying the product, the problem, and what should be done with the recalled product. We have found that not all of the recalls are picked up by the news media as they occur. This monthly summary will give you the opportunity to review all of the children’s product safety recalls for the past month. If you are interested in a complete text of the recall, double click on the hyperlink at the end of the recall description. This will direct you to the recall notice located on the CPSC website. Hallee Recalls Bed Canopies Due to Entanglement and Strangulation Hazards (17-701) FULLBEAUTY Brands Recalls Children’s Nightgowns Due to Violation of Federal Flammability Standard (17-702) Summer Infant Recalls Infant Bath Tubs Due to Risk of Impact Injury and Drowning (17-707) Mamas & Papas Recalls Armadillo Strollers Due to Fall Hazard (17-010) Roylco Recalls Educational Light Cubes Due to Fire Hazard WISCONSIN (17-012) DEPARTMENT OF Chimparoo Baby Carriers by L’echarpe Porte-bonheaur AGRICULTURE, Recalled Due to Fall Hazard (17-014) Target Recalls Halloween LED Gel Clings Due to Choking and TRADE AND Button Battery Ingestion Hazards (17-020) CONSUMER Fiddle Diddles Recalls Car Seat Strap Systems Due to PROTECTION Choking Hazard (17-705) If you would like to sign up for the Keep Your Kids Safe newsletter, please subscribe at https://public.govdelivery.com/accounts/WIDATCP/subscriber/new?topic_id OFFICE LOCATION =WIDATCP_161 or contact Bobbi Erb at (608) 224-4955 or [email protected]. -

Product Recalls Conceptualized As Social Dilemmas
PRODUCT RECALLS CONCEPTUALIZED AS SOCIAL DILEMMAS By SKYLER MASAJI KING A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY WASHINGTON STATE UNIVERSITY Carson College of Business MAY 2016 © Copyright by SKYLER MASAJI KING, 2016 All Rights Reserved © Copyright by SKYLER MASAJI KING, 2016 All Rights Reserved To the Faculty of Washington State University: The members of the Committee appointed to examine the dissertation of SKYLER MASAJI KING find it satisfactory and recommend that it be accepted. ___________________________________ Jeff Joireman, Ph.D., Chair ___________________________________ Andrew Perkins, Ph.D. ___________________________________ Joyce Ehrlinger, Ph.D. ii ACKNOWLEDGEMENT Throughout my time here at Washington State University, I have had the opportunity to meet exceptional scholars. First and foremost I would like to thank Dr. Jeff Joireman for his mentorship and guidance from day one. I could not have asked for a better mentor and friend throughout my time here. He has offered caring support throughout the program, allowing me to make and learn from my mistakes and never letting me take the easy way out through any process or project. Dr. Joireman, you have been a dedicated mentor and I sincerely hope to not only grow into a successful scholar like you are, but also grow into the type of person you are. I would also like to thank Drs. Andrew Perkins and Joyce Ehrlinger for the great resource they have been throughout the dissertation process. I feel very fortunate to have learned from them. Their academic pedigree is truly amazing. Additionally, their enthusiasm for research and helping students is second to none and I hope to emulate their knowledge and work ethic throughout my career in academia. -

Gossip, Exclusion, Competition, and Spite: a Look Below the Glass Ceiling at Female-To-Female Communication Habits in the Workplace
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 5-2013 Gossip, Exclusion, Competition, and Spite: A Look Below the Glass Ceiling at Female-to-Female Communication Habits in the Workplace Katelyn Elizabeth Brownlee [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Part of the Organizational Communication Commons Recommended Citation Brownlee, Katelyn Elizabeth, "Gossip, Exclusion, Competition, and Spite: A Look Below the Glass Ceiling at Female-to-Female Communication Habits in the Workplace. " Master's Thesis, University of Tennessee, 2013. https://trace.tennessee.edu/utk_gradthes/1597 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Katelyn Elizabeth Brownlee entitled "Gossip, Exclusion, Competition, and Spite: A Look Below the Glass Ceiling at Female-to-Female Communication Habits in the Workplace." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Master of Science, with a major in Communication and Information. Michelle Violanti, Major Professor We have read this thesis -

The Office” Sample Script
“THE OFFICE” SAMPLE SCRIPT “The Masseuse” by John Chang [email protected] FADE IN: INT. OFFICE – MORNING MICHAEL enters and stops by PAM’S desk. MICHAEL Morning, Pam. Did you catch the ‘L Word’ last night? PAM No. I missed it. MICHAEL It was a great episode. Tim found out that Jenny was cheating on him with Marina, and Dana and Lara broke up. But the whole thing was totally unbelievable. PAM Why? MICHAEL Because. There’s no way that lesbians are that hot in real life. I know that we all have our fantasies about a pair of hot lesbian chicks making out with each other, but that’s just not how it is in the real world. PAM Um, o-kay. MICHAEL I mean, seriously, Pam. There’s no way in a million years that a smoking hot lesbian babe would come up to you and ask you out on a date. It just wouldn’t happen. I mean, I’m sure you must be very attractive to plenty of lesbians out there, but let’s face facts: they don’t look like Jennifer Beals, they look like Rosie O’Donnell. 2 MICHAEL (cont’d) That’s why the ‘L Word’ is just a TV show, and this is real life. And Pam, for what it’s worth, if you were a lesbian, you’d be one of the hotter ones. PAM Um, thanks. As Michael heads for his office, Pam turns to the camera. Her expression asks, “Did he just say that?” END TEASER INT. OFFICE - DAY It’s business as usual, when the entrance of an extremely attractive young woman (MARCI) interrupts the office’s normal placid calm. -

THE WESTFIELD LEADER the Leading and Most Widely Circulated Weekly Xeuspaper in Union County
THE WESTFIELD LEADER The Leading and Most Widely Circulated Weekly Xeuspaper In Union County I'ulillahed 28 Pages—15 Cents EIGHl WESTFIELD, KEW JERSEY, THURSDAY, DECEMBER 21. 1978 ICvery Thumday T ^e Drinking Arrests Triple in '78 $15.5 Million School In a joint effort this week teenage arrests related to school Christmas vacation, grounds, are used as an area Parents must know. too. to reduce the rising in- alcoholic consumption. To parents are urged to give where teenagers gather and there have been incidents of cidence of teenage date, for this year, the special attention to teenage have drinking and smoking students bringing alcoholic alcoholism, the Children. number has risen to 106 parties, remembering that • marijuanai parties. As a beverages to school. The Youth and Recreation arrests, and the year is not the use of alcohol by minors display of cooperation to law is very explicit that no Budget Anticipated Committee of P-T Council. over Included in these is not only illegal, but ex- combat teenage drinking, student, regardless uf age. working with the Westfield statistics are children ages tremely dangerous to their marijuana usage and may use alcoholic Police Department, 13 through 17. Most of these young bodies. Police vandalism, the Westfield beverages while attending Indications of a 1979-80 elementary advanced is not bad," he said. year. released the following in- youngsters were so in-statistics reveal, also an Board of Education gave school, or any school func- school budget in the neigh- learning centers for above The -

Casino Skills for Working with Others Participant Manual: Edition 1.0
Casino Skills for Working with Others Participant Manual: Edition 1.0 Casino Skills for Working with Others: Essential Skills for the Gaming Industry Produced By: The Canadian Gaming Centre of Excellence (An operating division of The Manitoba Lotteries Corporation) 983 St. James Street Winnipeg, Manitoba R3H 0X2 (204) 957-2504 ext. 8498 e-mail: [email protected] Contact: Judith Hayes, Director Dayna Hinkel, Business Development Officer © 2010 Canadian Gaming Centre of Excellence (CGCE). All rights reserved. All material in this manual is the property of the CGCE. This publication, or any part thereof, may be used free of charge and permission is granted to reproduce for personal or educational use only. Commercial copying or selling is prohibited. In all cases this copyright notice must remain intact. Special Thanks: The Canadian Gaming Centre of Excellence would like to acknowledge the assistance of the following organizations and individuals in producing this manual: Aseneskak Casino Judy Goodridge, Chief Operating Officer Eleanor Gabriel, Human Resources Delores Lavallee, Manager, Human Resources Atlantic Lottery Corporation Alison Stultz, Director Organizational Development British Columbia Lottery Corporation: Mitch Romanchook, Manager Technical Services Talent Management – Human Resources Canadian Gaming Association Paul Burns, Vice President of Public Affairs Casino Rama Debra Pratt, Chief People Officer Century Casino & Hotel Nicole Jofre, Human Resources Great Canadian Gaming Corporation Sally Hart, Executive Director Human Resources -

ACEC Guide to Returning to the Office and the Job Site
ACEC Guide to Returning to the Office and the Job Site Member Firms across the country are facing the challenge of returning to the workplace and the job site during the COVID-19 pandemic. The purpose of this resource is to help your firm respond to this challenge safely while managing legal risk. The Guide is divided into five sections: (1) Planning the Return; (2) Health Policy and Procedures; (3) Returning to the Office; (4) Returning to the Job Site; and (5) links to additional COVID-19 resources. It incorporates information from a number of sources, including the CDC, OSHA, FDA, private sector organizations both in and outside the engineering industry, and legal counsel. We hope the Guide will help your firm to navigate the “new normal” of working in the midst of an ongoing public health emergency. freepik.com www.acec.org #RescueRecoverRebuild 1 Table of Contents Section 1: Planning the Return Appoint Return to Work Team.................................................................................... 3 Determine Transition Plan..........................................................................................3 Determine Which Shelter-in-Place Laws and Orders Apply........................................3 Identify Who Returns to the Workplace/Job Site and When...................................... 3 Develop and Train in Workplace Policies and Other Practices.................................. 4 Consider Administrative Measures.............................................................................4 Transition from Telework -

The Influence of Negative Workplace Gossip on Knowledge Sharing
sustainability Article The Influence of Negative Workplace Gossip on Knowledge Sharing: Insight from the Cognitive Dissonance Perspective Xiaolei Zou, Xiaoxi Chen * , Fengling Chen, Chuxin Luo and Hongyan Liu * School of Management, Jinan University, Guangzhou 510632, China; [email protected] (X.Z.); [email protected] (F.C.); [email protected] (C.L.) * Correspondence: [email protected] (X.C.); [email protected] (H.L.) Received: 4 March 2020; Accepted: 15 April 2020; Published: 17 April 2020 Abstract: Increasing attention is drawn to the effect of workplace gossip on the organization. Negative workplace gossip is a negative evaluation of others behind their back in the workplace. Based on the cognitive dissonance theory, the study explored the relationship between negative workplace gossip and knowledge sharing, through the mediation of organizational trust and the moderation of self-efficacy. The regression results of a two-stage questionnaire survey on 173 Chinese employees suggested that negative workplace gossip negatively influenced employees’ knowledge sharing through organizational trust. Additionally, findings also showed that self-efficacy moderated the mediation of organizational trust in the relationship between negative workplace gossip and knowledge sharing. This research provided a new theoretical perspective on the impact of workplace gossip, which has management implications for informal communication and team-building. Keywords: workplace gossip; knowledge sharing; organizational trust; self-efficacy 1. Introduction Workplace gossip is defined as informal conversation or evaluation (i.e., positive or negative) about a member beyond the person’s hearing [1], typically involving unproven details. Just as a famous proverb says, ‘Good news never goes beyond the gate, while bad news spreads far and wide’, gossip spreads rapidly and influences broadly. -
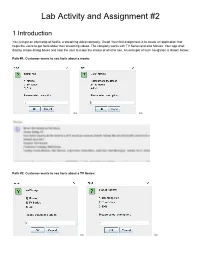
Lab Activity and Assignment #2
Lab Activity and Assignment #2 1 Introduction You just got an internship at Netfliz, a streaming video company. Great! Your first assignment is to create an application that helps the users to get facts about their streaming videos. The company works with TV Series and also Movies. Your app shall display simple dialog boxes and help the user to make the choice of what to see. An example of such navigation is shown below: Path #1: Customer wants to see facts about a movie: >> >> Path #2: Customer wants to see facts about a TV Series: >> >> >> >> Your app shall read the facts about a Movie or a TV Show from text files (in some other course you will learn how to retrieve this information from a database). They are provided at the end of this document. As part of your lab, you should be creating all the classes up to Section 3 (inclusive). As part of your lab you should be creating the main Netfliz App and making sure that your code does as shown in the figures above. The Assignment is due on March 8th. By doing this activity, you should be practicing the concept and application of the following Java OOP concepts Class Fields Class Methods Getter methods Setter methods encapsulation Lists String class Split methods Reading text Files Scanner class toString method Override superclass methods Scanner Class JOptionPane Super-class sub-class Inheritance polymorphism Class Object Class Private methods Public methods FOR loops WHILE Loops Aggregation Constructors Extending Super StringBuilder Variables IF statements User Input And much more.. -

Product Recalls, Imperfect Information, and Spillover Effects: Lessons from the Consumer Response to the 2007 Toy Recalls
NBER WORKING PAPER SERIES PRODUCT RECALLS, IMPERFECT INFORMATION, AND SPILLOVER EFFECTS: LESSONS FROM THE CONSUMER RESPONSE TO THE 2007 TOY RECALLS Seth M. Freedman Melissa Schettini Kearney Mara Lederman Working Paper 15183 http://www.nber.org/papers/w15183 NATIONAL BUREAU OF ECONOMIC RESEARCH 1050 Massachusetts Avenue Cambridge, MA 02138 July 2009 We gratefully acknowledge the helpful comments of Severin Borenstein, Jonathan Guryan, Judy Hellerstein, Ginger Jin, Arik Levinson, Soohyung Lee, Nuno Limao, and Abigail Wozniak as well as seminar participants at the University of Maryland, Rotman School of Management, the Energy Institute at U.C. Berkeley, Georgetown, UC-Davis, UC-Irvine, and University of Michigan. We thank Danny Kim at NPD for answering our questions about the toy sales data and Kevin Mak at the Rotman Finance Lab for his assistance with assembling the stock price data. Molly Reckson provided capable research assistance. Financial support from the AIC Institute for Corporate Citizenship at the Rotman School of Management is gratefully acknowledged. The views expressed herein are those of the author(s) and do not necessarily reflect the views of the National Bureau of Economic Research. NBER working papers are circulated for discussion and comment purposes. They have not been peer- reviewed or been subject to the review by the NBER Board of Directors that accompanies official NBER publications. © 2009 by Seth M. Freedman, Melissa Schettini Kearney, and Mara Lederman. All rights reserved. Short sections of text, not to exceed two paragraphs, may be quoted without explicit permission provided that full credit, including © notice, is given to the source. Product Recalls, Imperfect Information, and Spillover Effects: ¸˛Lessons from the Consumer Response to the 2007 Toy Recalls Seth M. -

Product Durability and Life
A111Q3 Qtbbfll \ V) NBS SPECIAL PUBLICATION 514 U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards Product Durability and Life MFPG 27th Meeting WATIONAL BURBAtJ OF STANDARDS JjIBflARY MFPG Product Durability and Life Proceedings of the 27th Meeting of the Mechanical Failures Prevention Group, held at the National Bureau of Standards, Gaithersburg, Maryland, November 1-3, 1977 Edited by . T. Robert Shives and William A. Willard National Measurement Laboratory National Bureau of Standards Washington, DC 20234 The 27th meeting of the MFPG and these proceedings were sponsored by the Institute for Materials Research and the Cen- ter for Consumer Product Technology of the National Bureau of Standards, Washington, DC 20234; the Office of Naval Research, Department of the Navy, Arlington, VA 22217; the Naval Air Development Center, Department of the Navy, Warminster, PA 18974; the National Aeronautics and Space Administration, Goddard Space Flight Center, Greenbelt, MD 20771; and the Department of Energy-Fossil Energy, Washington, DC 20545. This work was co-sponsored by the Institute for Materials Research prior to the NBS reorganization effective April 9, 1978. The Institute for Materials Research is now included in the National Measurement Laboratory of NBS. * Qin U.S. DEPARTMENT OF COMMERCE, Juanita M. Kreps, Secretary Dr. Sidney Harman, Under Secretary Jordan J. Baruch, Assistant Secretary for Science and Technology NATIONAL BUREAU OF STANDARDS, Ernest Annbler, Director Issued May 1978 Library of Congress Cataloging in Publication Data Mechanical Failures Prevention Group. MFPG, product durability and life. (National Bureau of Standards Special publication ; 514) "Sponsored by the Institute for Materials Research . [et asl.]." Supt.ofDocs.no.: C13.