Influence of Erf3a/GSPT1 Gene Expression on Susceptibility to Carcinogenesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Manuscript for Proximity Interactome Analysis of Lassa
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.16.452739; this version posted July 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Main Manuscript for 2 Proximity interactome analysis of Lassa polymerase reveals 3 eRF3a/GSPT1 as a druggable target for host directed antivirals. 4 Jingru Fang1,2, Colette Pietzsch3,4, George Tsaprailis5, Gogce Crynen5, Kelvin Frank Cho6, Alice 5 Y. Ting7,8, Alexander Bukreyev3,4,9, Erica Ollmann Saphire2* and Juan Carlos de la Torre1* 6 1 Department of Immunology and Microbiology, Scripps Research, La Jolla CA 92037 7 2La Jolla Institute for Immunology, La Jolla CA 92037 8 3Department of Pathology, University of Texas Medical Branch, Galveston, TX 77550 9 4Galveston National Laboratory, University of Texas Medical Branch, Galveston, TX 77550 10 5Proteomics Core, Scripps Research, Jupiter, FL 33458 11 6Cancer Biology Program, Stanford University, Stanford, CA 94305 12 7Department of Genetics, Department of Biology and Department of Chemistry, Stanford 13 University, Stanford, CA 94305 14 8Chan Zuckerberg Biohub, San Francisco, CA 94158 15 9Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, 16 TX 77550 17 *Erica Ollmann Saphire; Juan Carlos de la Torre. 18 Email: [email protected] and [email protected] 19 Author Contributions: J.F., E.O.S., and J.C.T designed research; J.F. performed all non-BSL4 20 experiments and analyzed data; C.P. -

Functional Interactomes of the Ebola Virus Polymerase Identified by Proximity Proteomics 2 in the Context of Viral Replication
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.20.453153; this version posted July 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Functional interactomes of the Ebola virus polymerase identified by proximity proteomics 2 in the context of viral replication 3 Jingru Fang1,2, Colette Pietzsch3,4, George Tsaprailis5, Gogce Crynen6, Kelvin Frank Cho7, Alice 4 Y. Ting8,9, Alexander Bukreyev3,4,10*, Juan Carlos de la Torre2*, Erica Ollmann Saphire1* 5 1La Jolla Institute for Immunology, La Jolla CA 92037 6 2Department of Immunology and Microbiology, Scripps Research, La Jolla CA 92037 7 3Department of Pathology, University of Texas Medical Branch, Galveston, TX 77550 8 4Galveston National Laboratory, University of Texas Medical Branch, Galveston, TX 77550 9 5Proteomics Core, Scripps Research, Jupiter, FL 33458 10 6Bioinformatics and Statistics Core, Scripps Research, Jupiter, FL 33458 11 7Cancer Biology Program, Stanford University, Stanford, CA 94305 12 8Department of Genetics, Department of Biology and Department of Chemistry, Stanford 13 University, Stanford, CA 94305 14 9Chan Zuckerberg Biohub, San Francisco, CA 94158 15 10 Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, 16 TX 77550 17 Correspondence: [email protected], [email protected], [email protected] 18 Lead contact: [email protected] 19 SUMMARY 20 Ebola virus (EBOV) critically depends on the viral polymerase to replicate and transcribe the viral 21 RNA genome. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

Uncovering RNA Binding Proteins Associated with Age and Gender During Liver Maturation
OPEN Uncovering RNA binding proteins SUBJECT AREAS: associated with age and gender during GENOME-WIDE ASSOCIATION STUDIES liver maturation REGULATORY NETWORKS Praneet Chaturvedi1, Yaseswini Neelamraju1, Waqar Arif2, Auinash Kalsotra2 & Sarath Chandra Janga1,3,4 AGEING GENE REGULATORY NETWORKS 1Department of BioHealth Informatics, School of Informatics and Computing, Indiana University Purdue University, 719 Indiana Ave Ste 319, Walker Plaza Building, Indianapolis, Indiana 46202, 2Departments of Biochemistry and Medical Biochemistry, University Received of Illinois, Urbana-Champaign, Illinois 61801, USA, 3Center for Computational Biology and Bioinformatics, Indiana University 11 December 2014 School of Medicine, 5021 Health Information and Translational Sciences (HITS), 410 West 10th Street, Indianapolis, Indiana, 46202, 4Department of Medical and Molecular Genetics, Indiana University School of Medicine, Medical Research and Library Accepted Building, 975 West Walnut Street, Indianapolis, Indiana, 46202. 9 March 2015 Published In the present study, we perform an association analysis focusing on the expression changes of 1344 RNA 31 March 2015 Binding proteins (RBPs) as a function of age and gender in human liver. We identify 88 and 45 RBPs to be significantly associated with age and gender respectively. Experimental verification of several of the predicted associations in mice confirmed our findings. Our results suggest that a small fraction of the gender-associated RBPs (,40%) are expressed higher in males than females. Altogether, these observations Correspondence and show that several of these RBPs are important and conserved regulators in maintaining liver function. requests for materials Further analysis of the protein interaction network of RBPs associated with age and gender based on the should be addressed to centrality measures like degree, betweenness and closeness revealed that several of these RBPs might be S.C.J. -

Nahar Et Al. Supplementary Figure 1 (%) 10
A (%) 5,000 4,000 3,000 hADSC 2,000 mADSC 1,000 0 day0 day3 day5 B CD90 CD44 hADSC 600µm 600µm CD90.2 CD44 mADSC 600µm 600µm Nahar et al. Supplementary Figure 1 (%) 10 0 2 4 6 8 biological adhesion biological regulation cell killing cellular process developme ntal proce ss establishment of localization Biological Process Biological growth immune system process localization locom otion metabolic process multi-organism process multicellular organismal process pigmentation reproduction reproductive process hADSC response to stimulus rhythmic process viral process Golgi apparatus mADSC cytoplasm cytoskeleton Cellular Component endoplasmic reticulum endosome extracellular region Both hADSC and mADSC and hADSC Both Nahar intracellular organelle membrane mitochondrion nucleus et al. organelle membrane organelle part plasma membrane ribosome Supplementary antioxidant activity transport binding catalytic activity obsolete chaperone regulator activity chemoattractant activity Molecular Function Molecular chemorepellent activity electron carrier activity enzyme regulator activity Figure metallochaperone activity molecular function molecular transducer activity motor activity 2 nutrient reservoir activity ( protein tag Figure structural molecule activity obsolete transcription regulator activity translation regulator activity 4) transporter activity Supplementary Table 1. Identification of endogenous proteins contained both hADSC and mADSC. a UniProt/SWISS- Biological Process Cellular Component Molecular Function emPAI PROT ID Alternate ID biological -

Mrna Expression in Human Leiomyoma and Eker Rats As Measured by Microarray Analysis
Table 3S: mRNA Expression in Human Leiomyoma and Eker Rats as Measured by Microarray Analysis Human_avg Rat_avg_ PENG_ Entrez. Human_ log2_ log2_ RAPAMYCIN Gene.Symbol Gene.ID Gene Description avg_tstat Human_FDR foldChange Rat_avg_tstat Rat_FDR foldChange _DN A1BG 1 alpha-1-B glycoprotein 4.982 9.52E-05 0.68 -0.8346 0.4639 -0.38 A1CF 29974 APOBEC1 complementation factor -0.08024 0.9541 -0.02 0.9141 0.421 0.10 A2BP1 54715 ataxin 2-binding protein 1 2.811 0.01093 0.65 0.07114 0.954 -0.01 A2LD1 87769 AIG2-like domain 1 -0.3033 0.8056 -0.09 -3.365 0.005704 -0.42 A2M 2 alpha-2-macroglobulin -0.8113 0.4691 -0.03 6.02 0 1.75 A4GALT 53947 alpha 1,4-galactosyltransferase 0.4383 0.7128 0.11 6.304 0 2.30 AACS 65985 acetoacetyl-CoA synthetase 0.3595 0.7664 0.03 3.534 0.00388 0.38 AADAC 13 arylacetamide deacetylase (esterase) 0.569 0.6216 0.16 0.005588 0.9968 0.00 AADAT 51166 aminoadipate aminotransferase -0.9577 0.3876 -0.11 0.8123 0.4752 0.24 AAK1 22848 AP2 associated kinase 1 -1.261 0.2505 -0.25 0.8232 0.4689 0.12 AAMP 14 angio-associated, migratory cell protein 0.873 0.4351 0.07 1.656 0.1476 0.06 AANAT 15 arylalkylamine N-acetyltransferase -0.3998 0.7394 -0.08 0.8486 0.456 0.18 AARS 16 alanyl-tRNA synthetase 5.517 0 0.34 8.616 0 0.69 AARS2 57505 alanyl-tRNA synthetase 2, mitochondrial (putative) 1.701 0.1158 0.35 0.5011 0.6622 0.07 AARSD1 80755 alanyl-tRNA synthetase domain containing 1 4.403 9.52E-05 0.52 1.279 0.2609 0.13 AASDH 132949 aminoadipate-semialdehyde dehydrogenase -0.8921 0.4247 -0.12 -2.564 0.02993 -0.32 AASDHPPT 60496 aminoadipate-semialdehyde -

The Effects of GSPT1 Degradation on Serum Calcium, Parathyroid Hormone, and Fibroblast Growth Factor 23 Concentrations in Human Cereblon Knock-In Mice
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2020 The Effects of GSPT1 Degradation on Serum Calcium, Parathyroid Hormone, and Fibroblast Growth Factor 23 Concentrations in Human Cereblon Knock-in Mice Kamran Ghoreishi Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Other Medicine and Health Sciences Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/6490 This Dissertation is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. The Effects of GSPT1 Degradation on Serum Calcium, Parathyroid Hormone, and Fibroblast Growth Factor 23 Concentrations in Human Cereblon Knock-in Mice by Kamran Ghoreishi Bachelor of Science in Microbiology and Clinical Laboratory Sciences, San Diego State University, 1995 Master of Science in Public Health (Toxicology), San Diego State University, 2003 Master of Science in Regulatory Affairs, San Diego State University, 2013 A dissertation submitted in the partial fulfillment of the requirements for the degree of Doctor of Philosophy at Virginia Commonwealth University. Virginia Commonwealth University, 2020 Advisor: William J. Korzun, Ph.D. Associate Professor Department of Clinical Laboratory Sciences Virginia Commonwealth University Richmond, Virginia November 19, 2020 ACKNOWLEDGEMENT Firstly, I would like to express my sincere gratitude to my advisor Dr. William Korzun for the continuous support and guidance of my research, which incented me to widen my research from various perspectives. My sincere thanks also go to Dr. -
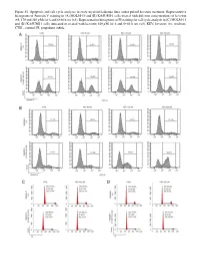
Figure S1. Apoptosis and Cell Cycle Analyses in Acute Myeloid Leukemia Lines Under Pulsed Kevetrin Treatment
Figure S1. Apoptosis and cell cycle analyses in acute myeloid leukemia lines under pulsed kevetrin treatment. Representative histograms of Annexin V staining in (A) MOLM‑13 and (B) KASUMI‑1 cells treated with different concentrations of kevetrin (85, 170 and 340 µM) for 6 and 6+66 h wo (x3). Representative histograms of PI staining for cell cycle analysis in (C) MOLM‑13 and (D) KASUMI‑1 cells untreated or treated with kevetrin 340 µM for 6 and 6+66 h wo (x3). KEV, kevetrin; wo, washout; CTRL, control; PI, propidium iodide. Figure S2. Viability of acute myeloid leukemia cell lines after 72 h of kevetrin treatment. Viability of OCI‑AML3, MOLM‑13, KASUMI‑1 and NOMO‑1 cell lines treated with different concentrations of kevetrin (85, 170 and 340 µM) for 72 h. Values represent the mean ± standard deviation of three biological replicates. **P<0.01, ***P<0.001. Figure S3. Apoptosis analysis in acute myeloid leukemia cell lines under 24 h of continuous kevetrin treatment. Representative histograms of Annexin V staining in OCI‑AML3, MOLM‑13, KASUMI‑1 and NOMO‑1 cells treated with different concentra‑ tions of kevetrin (85, 170 and 340 µM) for 24 h. KEV, kevetrin; CTRL, control. Figure S4. Apoptosis analysis in acute myeloid leukemia cell lines under 48 h of continuous kevetrin treatment. Representative histograms of Annexin V staining in OCI‑AML3, MOLM‑13, KASUMI‑1 and NOMO‑1 cells treated with different concentra‑ tions of kevetrin (85, 170 and 340 µM) for 48 h. KEV, kevetrin; CTRL, control. Figure S5. Apoptosis analyses in acute myeloid leukemia cell lines under continuous kevetrin treatment. -
Supplementary Materials For
Supplementary Materials for Identification of candidate biomarkers for Idiopathic Thrombocytopenic Purpura by Bioinformatics Analysis of Microarray Data Samira Gilanchi, Hakimeh Zali*, Mohammad Faranoush*, Mostafa Rezaei Tavirani, Keyvan Shahriary and Mahyar Daskareh To whom correspondence should be addressed. E-mail [email protected]; [email protected] Volume 19, issue 4 (autumn 2020) This PDF file includes: Tables S1 to S8 Table S1. Up-regulated DEGs that obtain from analysis from 7 newly diagnosed ITP than 6 chronic ITP samples. In table M means log2FC, T is T statistics, P-values evaluated based on Benjamini false discovery rate. Top genes are selected for more valuation with P-value < 0.05, and B is B statistic. Up-regulated genes M T P-Value B 1 FNBP4 1.089255 9.645201 0.0067318 6.692063 2 G2E3 0.993745 7.38905 0.0306136 4.269833 3 SAR1B 1.523974 7.35994 0.0306136 4.233735 4 PER3 1.297768 7.261088 0.0306136 4.11015 5 MSI2 1.60714 7.23665 0.0306136 4.079358 6 CASK 1.854101 7.07773 0.0324118 3.876782 7 TM2D1 1.510449 7.006354 0.0326833 3.784474 8 RPRD1A 1.155052 6.971092 0.0326833 3.738565 9 SLC39A6 1.032869 6.868857 0.033521 3.604313 10 CAMTA1 2.258868 6.855413 0.033521 3.586531 11 PTAR1 1.602835 6.828607 0.033521 3.550986 12 LOC101929734 1.620399 6.561583 0.039368 3.190433 13 ADNP2 1.084491 6.519255 0.0400375 3.132189 14 ZNF224 1.234505 6.365865 0.0400375 2.918603 15 TUBE1 0.990095 6.354317 0.0400375 2.902363 16 GIGYF2 0.741529 6.353043 0.0400375 2.900571 17 ABCA5 1.293839 6.341226 0.0400375 2.883927 18 FBXW7 1.205229 6.303732 -

Supplementary Tables
Supplementary Tables Supplementary Table 1. Differentially methylated genes in correlation with their expression pattern in the A4 progression model A. Hypomethylated–upregulated Genes (n= 76) ALOX5 RRAD RTN4R DSCR6 FGFR3 HTR7 WNT3A POGK PLCD3 ALPPL2 RTEL1 SEMA3B DUSP5 FOSB ITGB4 MEST PPL PSMB8 ARHGEF4 BST2 SEMA7A SLC12A7 FOXQ1 KCTD12 LETM2 PRPH PXMP2 ARNTL2 CDH3 SHC2 SLC20A2 HSPA2 KIAA0182 LIMK2 NAB1 RASIP1 ASRGL1 CLDN3 DCBLD1 SNX10 SSH1 KREMEN2 LIPE NDRG2 ATF3 CLU DCHS1 SOD3 ST3GAL4 MAL LRRC1 NR3C2 ATP8B3 CYC1 DGCR8 EBAG9 SYNGR1 TYMS MCM2 NRG2 RHOF DAGLA DISP2 FAM19A5 TNNI3 UNC5B MYB PAK6 RIPK4 DAZAP1 DOCK3 FBXO6 HSPA4L WHSC1 PNMT PCDH1 B. Hypermethylated-downregulated Genes (n= 31) ARHGAP22 TNFSF9 KLF6 LRP8 NRP1 PAPSS2 SLC43A2 TBC1D16 ASB2 DZIP1 TPM1 MDGA1 NRP2 PIK3CD SMARCA2 TLL2 C18orf1 FBN1 LHFPL2 TRIO NTNG2 PTGIS SOCS2 TNFAIP8 DIXDC1 KIFC3 LMO1 NR3C1 ODZ3 PTPRM SYNPO Supplementary Table 2. Genes enriched for different histone methylation marks in A4 progression model identified through ChIP-on-chip a. H3K4me3 (n= 978) AATF C20orf149 CUL3 FOXP1 KATNA1 NEGR1 RAN SPIN2B ABCA7 C20orf52 CWF19L1 FRK KBTBD10 NEIL1 RANBP2 SPPL2A ABCC9 C21orf13-SH3BGR CXCL3 FSIP1 KBTBD6 NELF RAPGEF3 SPRY4 ABCG2 C21orf45 CYC1 FUK KCMF1 NFKB2 RARB SPRYD3 ABHD7 C22orf32 CYorf15A FXR2 KCNH7 NGDN RASAL2 SPTLC2 ACA15 C2orf18 DAXX FZD9 KCNMB4 NKAP RASD1 SRFBP1 ACA26 C2orf29 DAZ3 G6PD KCTD18 NKTR RASEF SRI ACA3 C2orf32 DBF4 GABPB2 KDELR2 NNT RASGRF1 SRM ACA48 C2orf55 DBF4B GABRA5 KIAA0100 NOL5A RASSF1 SSH2 ACAT1 C3orf44 DBI GADD45B KIAA0226 NOLC1 RASSF3 SSH3 ACSL5 -

Exome Sequencing Enhanced Package Department of Pathology and Laboratory Medicine Feb 2012 UCLA Molecular Diagnostics Laboratories Page:1
UCLA Health System Clinical Exome Sequencing Enhanced Package Department of Pathology and Laboratory Medicine Feb 2012 UCLA Molecular Diagnostics Laboratories Page:1 Gene_Symbol Total_coding_bp %_bp_>=10X Associated_Disease(OMIM) MARC1 1093 80% . MARCH1 1005 100% . MARC2 1797 92% . MARCH3 802 100% . MARCH4 1249 99% . MARCH5 861 96% . MARCH6 2907 100% . MARCH7 2161 100% . MARCH8 900 100% . MARCH9 1057 73% . MARCH10 2467 100% . MARCH11 1225 56% . SEPT1 1148 100% . SEPT2 1341 100% . SEPT3 1175 100% . SEPT4 1848 96% . SEPT5 1250 94% . SEPT6 1440 96% . SEPT7 1417 96% . SEPT8 1659 98% . SEPT9 2290 96% Hereditary Neuralgic Amyotrophy SEPT10 1605 98% . SEPT11 1334 98% . SEPT12 1113 100% . SEPT14 1335 100% . SEP15 518 100% . DEC1 229 100% . A1BG 1626 82% . A1CF 1956 100% . A2LD1 466 42% . A2M 4569 100% . A2ML1 4505 100% . UCLA Health System Clinical Exome Sequencing Enhanced Package Department of Pathology and Laboratory Medicine Feb 2012 UCLA Molecular Diagnostics Laboratories Page:2 Gene_Symbol Total_coding_bp %_bp_>=10X Associated_Disease(OMIM) A4GALT 1066 100% . A4GNT 1031 100% . AAAS 1705 100% Achalasia‐Addisonianism‐Alacrima Syndrome AACS 2091 94% . AADAC 1232 100% . AADACL2 1226 100% . AADACL3 1073 100% . AADACL4 1240 100% . AADAT 1342 97% . AAGAB 988 100% . AAK1 3095 100% . AAMP 1422 100% . AANAT 637 93% . AARS 3059 100% Charcot‐Marie‐Tooth Neuropathy Type 2 AARS 3059 100% Charcot‐Marie‐Tooth Neuropathy Type 2N AARS2 3050 100% . AARSD1 1902 98% . AASDH 3391 100% . AASDHPPT 954 100% . AASS 2873 100% Hyperlysinemia AATF 1731 99% . AATK 4181 78% . ABAT 1563 100% GABA‐Transaminase Deficiency ABCA1 6991 100% ABCA1‐Associated Familial High Density Lipoprotein Deficiency ABCA1 6991 100% Familial High Density Lipoprotein Deficiency ABCA1 6991 100% Tangier Disease ABCA10 4780 100% .