Rechargeable Batteries
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Experiment on Charging an Electric Vehicle Lifepo4 Battery After Over-Discharge
INTERNATIONAL SCIENTIFIC JOURNAL "MACHINES. TECHNOLOGIES. MATERIALS" WEB ISSN 1314-507X; PRINT ISSN 1313-0226 Experiment on charging an electric vehicle LiFePO4 battery after over-discharge Nikolay Pavlov*, Diana Dacova Technical University - Sofia, Bulgaria [email protected] Abstract: The efficiency and technical and economic properties of the electric cars depend mainly on the rechargeable traction battery. LiFePo4 batteries belongs to the lithium-ion type and has a number of advantages such as high capacity, long life cycle, resistance to fire at high temperatures or shock. They have safe and stable over-charging and over-discharging performances. This paper describes the process of charging the individual cells of an electric car battery after their over-discharge. Keywords: LITHIUM IRON PHOSPHATE (LiFePO4) BATTERY, OVER-DISCHARGE, CHARGING, ELECTRIC VEHICLE 1. Introduction the battery does not ignite, explode or smoke. The authors of this publication have also performed experiments on over-discharged Early vehicles were created to meet the transport needs in the cells with high-current charging, using the on-board charger of the settlements. Due to low speeds and low mileage, in the middle of electric car. In this experiment, some of the cells deformed the 19th and the beginning of the 20th century the use of electric (swollen). cars and cars with internal combustion engine was equal, but social In this work the process of charging the battery of an electric and technical factors gave an advantage in the development of cars car after over-discharging of the individual cells is described. [1]. The harmful effects of emissions from internal combustion Charge experiments of lithium iron phosphate (LiFePO4) battery engines, the reduction of fossil fuel resources and improvements in have been performed on an electric car. -

Lithium Phosphate Refining Vindicates Cathode Production with No Requirement for Lithium Hydroxide Or Carbonate
05 March 2019 Lithium phosphate refining vindicates cathode production with no requirement for lithium hydroxide or carbonate HIGHLIGHTS ▪ Lithium phosphate produced from Lithium Australia's SiLeach® Gen-2 Pilot Plant has been further refined using a simple, proprietary purification process. ▪ The resulting product has a significantly higher purity than that previously used to create lithium-iron-phosphate cells via Lithium Australia's 100%-owned VSPC cathode powder technology. ▪ The latest refining process paves the way for the production of very high-purity cathodes while bypassing the supply restrictions of lithium carbonates and hydroxides. ▪ High-quality lithium-ion batteries generated from mine waste are now a reality. Lithium Australia NL (ASX: LIT, or 'the Company') has successfully refined lithium phosphate (‘LP') generated by its SiLeach® Gen-2 Pilot Plant to create a very pure product that is suitable for the direct generation of lithium-iron-phosphate ('LFP') powders for the production of lithium-ion batteries ('LIBs'). The Company's production of LIBs using LP generated direct from mine waste was first reported on 21 November 2018. Batteries produced in that manner demonstrated outstanding potential, notwithstanding the fact that they were manufactured from unrefined LP. Now, further refining of the LP by way of a simple proprietary process (details of which will be provided once updated IP applications are in place) has reduced the concentrations of impurities such as potassium, sodium and sulphur by orders of magnitude, providing scope to further improve battery performance. The new LP refining stage fits seamlessly into the Company's SiLeach® process, developed to capitalise on the abundance of lithium micas (for which SiLeach® is ideally suited). -

~Ui&£R5itt! of J\Rij!Oua
Minerals and metals of increasing interest, rare and radioactive minerals Authors Moore, R.T. Rights Arizona Geological Survey. All rights reserved. Download date 06/10/2021 17:57:35 Link to Item http://hdl.handle.net/10150/629904 Vol. XXIV, No.4 October, 1953 ~ui&£r5itt! of J\rij!oua ~ul1etiu ARIZONA BUREAU OF MINES MINERALS AND METALS OF INCREASING INTEREST RARE AND RADIOACTIVE MINERALS By RICHARD T. MOORE ARIZONA BUREAU OF MINES MINERAL TECHNOLOGY SERIES No. 47 BULLETIN No. 163 THIRTY CENTS (Free to Residents of Arizona) PUBLISHED BY ~tti£ll~r5itt! of ~rh!Omt TUCSON, ARIZONA TABLE OF CONTENTS INTRODUCTION 5 Acknowledgments 5 General Features 5 BERYLLIUM 7 General Features 7 Beryllium Minerals 7 Beryl 7 Phenacite 8 Gadolinite 8 Helvite 8 Occurrence 8 Prices and Possible Buyers ,........................................ 8 LITHIUM 9 General Features 9 Lithium Minerals 9 Amblygonite 9 Spodumene 10 Lepidolite 10 Triphylite 10 Zinnwaldite 10 Occurrence 10 Prices and Possible Buyers 10 CESIUM AND RUBIDIUM 11 General Features 11 Cesium and Rubidium Minerals 11 Pollucite ..................•.........................................................................., 11 Occurrence 12 Prices and Producers 12 TITANIUM 12 General Features 12 Titanium Minerals 13 Rutile 13 Ilmenite 13 Sphene 13 Occurrence 13 Prices and Buyers 14 GALLIUM, GERMANIUM, INDIUM, AND THALLIUM 14 General Features 14 Gallium, Germanium, Indium and Thallium Minerals 15 Germanite 15 Lorandite 15 Hutchinsonite : 15 Vrbaite 15 Occurrence 15 Prices and Producers ~ 16 RHENIUM 16 -

Alkaline Process for Extracting Lithium from Spodumene
Alkaline Process for Extracting Lithium from Spodumene Paulo Braga1, Silvia França1, Reiner Neumann1, Mario Rodriguez2 and Gustavo Rosales2 1. Center for Mineral Technology (CETEM), Brazil 2. Laboratory of Extractive Metallurgy and Materials Synthesis, Universidad Nacional de Cuyo, Argentina ABSTRACT The growing market demand for lithium is mainly due to its use in the manufacture of batteries for electric or hybrid vehicles and portable equipment (cellphones, tablets, power tools, notebooks, etc.). Currently there is great interest in finding new sources of lithium and technologies for its utilization. Brazil has large lithium pegmatite reserves and spodumene is the most important commercially mined lithium mineral. There are two main routes to obtain lithium carbonate and lithium hydroxide from mineral concentrates. In the first - the acid process - the mineral concentrate is roasted, sulfated with sulfuric acid and leached with water. Soda ash is added to form lithium carbonate. In the second process - the alkaline one - the mineral concentrate is roasted with lime or limestone, forming a clinker which is leached with water, filtered and crystallized as lithium hydroxide monohydrate. In Brazil, only the acidic process is used, although lithium hydroxide is the product most in demand in the domestic market, used mainly by automotive lubricant manufacturers. Lithium hydroxide is the main product in the alkaline process. The processing of spodumene concentrate to produce lithium hydroxide via the alkaline process can provide advantages to the production process, especially by replacing expensive chemicals such as sulfuric acid and soda ash with products such as limestone or hydrated lime, which are produced domestically and have more affordable prices. -

Electron Transport and Ion Diffusivity Through the Solid
ELECTRON TRANSPORT AND ION DIFFUSIVITY THROUGH THE SOLID ELECTROLYTE INTERPHASE IN LITHIUM ION BATTERIES WITH SILICON ANODES A Dissertation by LAURA ELENA BENITEZ Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Chair of Committee, Jorge M. Seminario Committee Members, Jun Zou Perla B. Balbuena Jose Silva-Martinez Head of Department, Miroslav M. Begovic August 2017 Major Subject: Electrical Engineering Copyright 2017 Laura Elena Benitez ABSTRACT Lithium-ion batteries (LIB) are the best option among batteries for portable electronic, power tools, and electric vehicles due to their higher energy storage, higher power, and lighter weight than other battery technologies such as Ni-based or lead acid. However, Li-ion batteries still face challenges such as safety, life, performance, and cost. One way to contribute to the solutions of these challenges and, consequently, improve the performance of Li-ion cells is to develop and design more stable passivation films at the electrode-electrolyte interface. Therefore, having a better understanding of the molecular processes that lead to the nucleation, growth, structure and morphology, as well as the electron and ion transport properties of the solid electrolyte interphase (SEI) is highly important for the development of new or improved lithium-ion batteries. In this work, computational methods, which allow studying phenomena not easily observable with experimental techniques, are used to study the electron transfer characteristics and the lithium ion diffusivity of the materials found in the SEI film formed in LIB with silicon anodes. First, ab initio computational methods are used to study the electron transfer through selected finite models of SEI films formed at the anode-electrolyte interface. -

Light-Assisted Delithiation of Lithium Iron Phosphate Nanocrystals Towards Photo-Rechargeable Lithium Ion Batteries
ARTICLE Received 7 Nov 2016 | Accepted 17 Jan 2017 | Published 10 Apr 2017 DOI: 10.1038/ncomms14643 OPEN Light-assisted delithiation of lithium iron phosphate nanocrystals towards photo-rechargeable lithium ion batteries Andrea Paolella1,2, Cyril Faure1, Giovanni Bertoni3, Sergio Marras4, Abdelbast Guerfi1, Ali Darwiche1, Pierre Hovington1, Basile Commarieu1, Zhuoran Wang2, Mirko Prato4, Massimo Colombo4, Simone Monaco4, Wen Zhu1, Zimin Feng1, Ashok Vijh1, Chandramohan George5, George P. Demopoulos2, Michel Armand6 & Karim Zaghib1 Recently, intensive efforts are dedicated to convert and store the solar energy in a single device. Herein, dye-synthesized solar cell technology is combined with lithium-ion materials to investigate light-assisted battery charging. In particular we report the direct photo- oxidation of lithium iron phosphate nanocrystals in the presence of a dye as a hybrid photo-cathode in a two-electrode system, with lithium metal as anode and lithium hexafluorophosphate in carbonate-based electrolyte; a configuration corresponding to lithium ion battery charging. Dye-sensitization generates electron–hole pairs with the holes aiding the delithiation of lithium iron phosphate at the cathode and electrons utilized in the formation of a solid electrolyte interface at the anode via oxygen reduction. Lithium iron phosphate acts effectively as a reversible redox agent for the regeneration of the dye. Our findings provide possibilities in advancing the design principles for photo-rechargeable lithium ion batteries. 1 Institute de Recherche d-Hydro-Que´bec (IREQ), 1800 Boulevard Lionel Boulet, Varennes, Quebec, Canada J3X 1S1. 2 Department of Mining and Materials Engineering, McGill University, Wong Building, 3610 University Street, Montreal, Quebec, Canada H3A OC5. 3 IMEM-CNR, Parco Area delle Scienze 37/A, 43124 Parma, Italy. -

Preparation of Lifepo4/C Cathode Materials Via a Green Synthesis Route for Lithium-Ion Battery Applications
materials Article Preparation of LiFePO4/C Cathode Materials via a Green Synthesis Route for Lithium-Ion Battery Applications Rongyue Liu 1,2,*,†, Jianjun Chen 1,*, Zhiwen Li 1,3,†, Qing Ding 2, Xiaoshuai An 3, Yi Pan 2, Zhu Zheng 2, Minwei Yang 2 and Dongju Fu 1 1 Research Institute of Tsinghua University in Shenzhen, High-Tech Industry Park, Nanshan District, Shenzhen 518057, China; [email protected] (Z.L.); [email protected] (D.F.) 2 Shenzhen Institute of THz Technology and Innovation, Xixiang, Bao’an District, Shenzhen 518102, China; [email protected] (Q.D.); [email protected] (Y.P.); [email protected] (Z.Z.); [email protected] (M.Y.) 3 Shenzhen Graduate School, Harbin Institute of Technology, Shenzhen University Town, Xili, Shenzhen 518055, China; [email protected] * Correspondence: [email protected] (R.L.); [email protected] (J.C.) † The authors equally contributed to this work. Received: 12 October 2018; Accepted: 9 November 2018; Published: 12 November 2018 Abstract: In this work, LiFePO4/C composite were synthesized via a green route by using Iron (III) oxide (Fe2O3) nanoparticles, Lithium carbonate (Li2CO3), glucose powder and phosphoric acid (H3PO4) solution as raw materials. The reaction principles for the synthesis of LiFePO4/C composite were analyzed, suggesting that almost no wastewater and air polluted gases are discharged into the environment. The morphological, structural and compositional properties of the LiFePO4/C composite were characterized by X-ray diffraction (XRD), scanning electron microscope (SEM), transmission electron microscopy (TEM), Raman and X-ray photoelectron spectroscopy (XPS) spectra coupled with thermogravimetry/Differential scanning calorimetry (TG/DSC) thermal analysis in detail. -

INFLUENCE COMPARISON of PRECURSORS on Lifepo4/C CATHODE STRUCTURE for LITHIUM ION BATTERIES
JKPK (JURNAL KIMIA DAN PENDIDIKAN KIMIA), Vol. 5, No. 1, 2020 pp. 24-31 Chemistry Education Study Program, Universitas Sebelas Maret ISSN 2503-4146 https://jurnal.uns.ac.id/jkpk ISSN 2503-4154 (online) INFLUENCE COMPARISON OF PRECURSORS ON LiFePO4/C CATHODE STRUCTURE FOR LITHIUM ION BATTERIES Luthfi Mufidatul Hasanah, Cornelius Satria Yudha, Soraya Ulfa Muzayanha, Diajeng Putri Suciutami, Atika Aulia Novita Sari, Inayati, and Agus Purwanto* Postgraduate Chemical Engineering, Faculty of Engineering, Universitas Sebelas Maret Jl. Ir. Sutami No. 36 A, Surakarta 57126, Central Java, Indonesia * Correspondence: Tel/Fax (0271)-632112, email: [email protected] Received: May 07, 2019 Accepted: April 04, 2020 Online Published: April 30, 2020 DOI : 10.20961/jkpk.v5i1.29874 ABSTRACT Electricity is the most energy demanded in this era. Energy storage devices must be able to store long-term and portable. A lithium ion battery is a type of battery that has been occupied in a secondary battery market. Lithium iron phosphate / LiFePO4 is a type of cathode material in ion lithium batteries that is very well known for its environmental friendliness and low prices. LiFePO4/C powder can be obtained from the solid state method. In this study the variables used were the types of precursors : iron sulfate (FeSO4), iron oxalate (FeC2O4) and FeSO4+charcoal. Synthesis of LiFePO4/C powder using Li:Fe:P at 1:1:1 %mol. Based on the XRD results, LiFePO4/C from FeSO4+charcoal shows the LiFePO4/C peaks according to the JCPDS Card with slight impurities when compared to other precursors. XRD results of LiFePO4/C with precursors of FeSO4 or FeC2O4 shows more impurities peaks. -

The Lithium-Ion Battery Value Chain
THE LITHIUM-ION BATTERY VALUE CHAIN New Economy Opportunities for Australia Acknowledgment Austrade would like to express our appreciation to Future Smart Strategies, especially Howard Buckley, for his professional guidance, advice and assistance, with earlier versions of this report. We would also like to thank Adrian Griffin at Lithium Australia for his insights and constructive suggestions. And we would like to acknowledge the insights provided by Prabhav Sharma at McKinsey & Company. More broadly, we would like to thank the following companies and organisations for providing data and information that assisted our research: › Association of Mining and Exploration Australia (AMEC); › Geoscience Australia; › Albemarle; and › TianQi Australia. Disclaimer Copyright © Commonwealth of Australia 2018 This report has been prepared by the Commonwealth of Australia represented by the Australian Trade and Investment Commission (Austrade). The report is a general overview and is not intended to The material in this document is licensed under a Creative Commons provide exhaustive coverage of the topic. The information is made Attribution – 4.0 International licence, with the exception of: available on the understanding that the Commonwealth of Australia is • the Australian Trade and Investment Commission’s logo not providing professional advice. • any third party material While care has been taken to ensure the information in this report • any material protected by a trade mark is accurate, the Commonwealth does not accept any liability for any • any images and photographs. loss arising from reliance on the information, or from any error or More information on this CC BY licence is set out at the creative omission, in the report. -

New Advances in Lithium Ion Battery Fuel Gauging Final
New Advances in Lithium Ion Battery Monitoring Jörn Tinnemeyer Vice President - Research & Development Cadex Electronics Inc. 22000 Fraserwood Way, Richmond BC, Canada V6W 1J6 [email protected] or www.cadex.com Abstract: In the last decade, lithium ion batteries have Importantly, though, the praises of Li-ion batteries must dominated the market place: first being used in portable be tempered by several key disadvantages in using this consumer products, and now in more industrial and chemistry. Namely, they safely operate within a very transport-based applications. One necessary requirement limited range of conditions; that is, the manufacturers of of lithium ion batteries -- irrespective of the particular Li-ion battery packs must be vigilant when developing the application of interest -- is to gauge how much energy the protection circuitry and a safe milieu for the battery battery contains and how long a given application can (McDowall et al., 2007, Van Schalkwijk et al., 2002). run before the battery needs to be recharged. The precise monitoring and management of lithium ion batteries has The precise monitoring and management of Li-ion proven to be difficult to achieve, especially as the battery batteries also presents a noteworthy problem -- this starts to age. Here, I describe a novel patented approach problem being the focus of this article. Irrespective of the that Cadex Electronics Inc., is developing, which assesses application being considered, the end-user needs to know the state of charge and state of health of lithium ion when the battery is fully charged and when the battery batteries by directly measuring the concentration of lithium will run out of power. -

Thermally Modulated Lithium Iron Phosphate Batteries for Mass-Market Electric Vehicles
ARTICLES https://doi.org/10.1038/s41560-020-00757-7 Thermally modulated lithium iron phosphate batteries for mass-market electric vehicles Xiao-Guang Yang 1, Teng Liu1 and Chao-Yang Wang 1,2 ✉ The pursuit of energy density has driven electric vehicle (EV) batteries from using lithium iron phosphate (LFP) cathodes in early days to ternary layered oxides increasingly rich in nickel; however, it is impossible to forgo the LFP battery due to its unsurpassed safety, as well as its low cost and cobalt-free nature. Here we demonstrate a thermally modulated LFP battery to offer an adequate cruise range per charge that is extendable by 10 min recharge in all climates, essentially guaranteeing EVs that are free of range anxiety. Such a thermally modulated LFP battery designed to operate at a working temperature around 60 °C in any ambient condition promises to be a well-rounded powertrain for mass-market EVs. Furthermore, we reveal that the limited working time at the high temperature presents an opportunity to use graphite of low surface areas, thereby prospec- tively prolonging the EV lifespan to greater than two million miles. he transportation sector accounts for 29% of US greenhouse rise (peak temperature <79 °C) on nail penetration. A similar test gas emissions, 59% of which come from light-duty vehicles on a seven-cell NMC module caused fires in all cells with a peak making personal trips1. Powertrain electrification is a prom- temperature of 549 °C. Wang and colleagues22 compared the fire T 2 ising route for decarbonized transportation . In light of the Paris hazards of 80 Ah LFP and 50 Ah NMC cells by triggering thermal Agreement, many countries have announced plans to phase out runaway with a resistive heater and found that the NMC cell had internal combustion engine (ICE) vehicles and incentivize electric 2.9 times the heat release rate and three times the toxic CO release vehicles (EVs). -
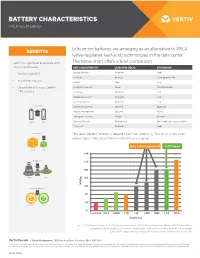
Battery Characteristics: VRLA Vs. Lithium
BATTERY CHARACTERISTICS VRLA vs Lithium-ion Lithium-ion batteries are emerging as an alternative to VRLA BENEFITS (valve-regulated lead-acid) technologies in the data center. Vertiv has significant experience with The below chart offers a brief comparison. lithium-ion batteries. KEY CHARACTERISTIC LEAD-ACID (VRLA) LITHIUM-ION* yyTesting since 2011 Energy Density Moderate High Lifespan Medium Long (approx. 4X) yy Installation success Weight High Low yy Compatible with many Liebert® Footprint required Large Small-Moderate UPS systems Recharge Moderate Fast Maintenance cost Moderate Low Cooling required Moderate Low Longer life Battery Monitoring Optional Optional Battery Management Optional Built-in x4 Transport concerns Flexible Ground VRLA LIB Disposal/Recycle Widespread Disposable, not yet recyclable First cost Moderate High Saves space * Batteries perform differently depending on their chemistry. This chart summarizes 70% various types. Note our preferences for critical use (gray). Data Center Approved Cell Phones VRLA LIB 280 Weighs less 240 60% 10 LB 4 LB 200 VRLA LIB 160 Wh/kg 120 Saves cooling cost 70% 80 kWh kWh 40 VRLA LIB 0 Lead Acid NiCd NiMH LTO LFP LMO NMC LCO NCA Chemistry Key: LTO (Lithium Titanate), LFP (Lithium Iron Phosphate), LMO (Lithium Manganese Oxide), NMC (Lithium Nickel Manganese Cobalt Oxide), LCO (Lithium Cobalt Oxide), NCA (Lithium Nickel Cobalt Aluminum Oxide). Chart data is copyrighted by BatteryUniversity.com and provided with permission. VertivCo.com | Vertiv Headquarters, 1050 Dearborn Drive, Columbus, OH, 43085, USA © 2017 Vertiv Co. All rights reserved. Vertiv, the Vertiv logo and Vertiv Liebert DSE are trademarks or registered trademarks of Vertiv Co. All other names and logos referred to are trade names, trademarks or registered trademarks of their respective owners.