Microrna-302D Targets IRF9 to Regulate the IFN-Induced Gene Expression in SLE
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Molecular Switch from STAT2-IRF9 to ISGF3 Underlies Interferon-Induced Gene Transcription
ARTICLE https://doi.org/10.1038/s41467-019-10970-y OPEN A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription Ekaterini Platanitis 1, Duygu Demiroz1,5, Anja Schneller1,5, Katrin Fischer1, Christophe Capelle1, Markus Hartl 1, Thomas Gossenreiter 1, Mathias Müller2, Maria Novatchkova3,4 & Thomas Decker 1 Cells maintain the balance between homeostasis and inflammation by adapting and inte- grating the activity of intracellular signaling cascades, including the JAK-STAT pathway. Our 1234567890():,; understanding of how a tailored switch from homeostasis to a strong receptor-dependent response is coordinated remains limited. Here, we use an integrated transcriptomic and proteomic approach to analyze transcription-factor binding, gene expression and in vivo proximity-dependent labelling of proteins in living cells under homeostatic and interferon (IFN)-induced conditions. We show that interferons (IFN) switch murine macrophages from resting-state to induced gene expression by alternating subunits of transcription factor ISGF3. Whereas preformed STAT2-IRF9 complexes control basal expression of IFN-induced genes (ISG), both type I IFN and IFN-γ cause promoter binding of a complete ISGF3 complex containing STAT1, STAT2 and IRF9. In contrast to the dogmatic view of ISGF3 formation in the cytoplasm, our results suggest a model wherein the assembly of the ISGF3 complex occurs on DNA. 1 Max Perutz Labs (MPL), University of Vienna, Vienna 1030, Austria. 2 Institute of Animal Breeding and Genetics, University of Veterinary Medicine Vienna, Vienna 1210, Austria. 3 Institute of Molecular Biotechnology of the Austrian Academy of Sciences (IMBA), Vienna 1030, Austria. 4 Research Institute of Molecular Pathology (IMP), Vienna Biocenter (VBC), Vienna 1030, Austria. -

Genome-Scale Identification of Transcription Factors That Mediate An
ARTICLE DOI: 10.1038/s41467-018-04406-2 OPEN Genome-scale identification of transcription factors that mediate an inflammatory network during breast cellular transformation Zhe Ji 1,2,4, Lizhi He1, Asaf Rotem1,2,5, Andreas Janzer1,6, Christine S. Cheng2,7, Aviv Regev2,3 & Kevin Struhl 1 Transient activation of Src oncoprotein in non-transformed, breast epithelial cells can initiate an epigenetic switch to the stably transformed state via a positive feedback loop that involves 1234567890():,; the inflammatory transcription factors STAT3 and NF-κB. Here, we develop an experimental and computational pipeline that includes 1) a Bayesian network model (AccessTF) that accurately predicts protein-bound DNA sequence motifs based on chromatin accessibility, and 2) a scoring system (TFScore) that rank-orders transcription factors as candidates for being important for a biological process. Genetic experiments validate TFScore and suggest that more than 40 transcription factors contribute to the oncogenic state in this model. Interestingly, individual depletion of several of these factors results in similar transcriptional profiles, indicating that a complex and interconnected transcriptional network promotes a stable oncogenic state. The combined experimental and computational pipeline represents a general approach to comprehensively identify transcriptional regulators important for a biological process. 1 Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115, USA. 2 Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA. 3 Department of Biology, Howard Hughes Medical Institute and David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 20140, USA. 4Present address: Department of Pharmacology and Biomedical Engineering, Northwestern University, Evanston 60611 IL, USA. -

Figure S1. Basic Information of RNA-Seq Results. (A) Bar Plot of Reads Component for Each Sample
Figure S1. Basic information of RNA-seq results. (A) Bar plot of reads component for each sample. (B) Dot plot shows the principal component analysis (PCA) of each sample. (C) Venn diagram of DEGs for three time points, the overlap part of the circles represents common differentially expressed genes between combinations. Figure S2. Scatter plot of DEGs for each time point. The X and Y axes represent the logarithmic value of gene expression. Red represents up-regulated DEG, blue represents down-regulated DEG, and gray represents non-DEG. Table S1. Primers used for quantitative real-time PCR analysis of DEGs. Gene Primer Sequence Forward 5’-CTACGAGTGGATGGTCAAGAGC-3’ FOXO1 Reverse 5’-CCAGTTCCTTCATTCTGCACACG-3’ Forward 5’-GACGTCCGGCATCAGAGAAA-3’ IRS2 Reverse 5’-TCCACGGCTAATCGTCACAG-3’ Forward 5’-CACAACCAGGACCTCACACC-3’ IRS1 Reverse 5’-CTTGGCACGATAGAGAGCGT-3’ Forward 5’-AGGATACCACTCCCAACAGACCT-3’ IL6 Reverse 5’-CAAGTGCATCATCGTTGTTCATAC-3’ Forward 5’-TCACGTTGTACGCAGCTACC-3’ CCL5 Reverse 5’-CAGTCCTCTTACAGCCTTTGG-3’ Forward 5’-CTGTGCAGCCGCAGTGCCTACC-3’ BMP7 Reverse 5’-ATCCCTCCCCACCCCACCATCT-3’ Forward 5’-CTCTCCCCCTCGACTTCTGA-3’ BCL2 Reverse 5’-AGTCACGCGGAACACTTGAT-3’ Forward 5’-CTGTCGAACACAGTGGTACCTG-3’ FGF7 Reverse 5’-CCAACTGCCACTGTCCTGATTTC-3’ Forward 5’-GGGAGCCAAAAGGGTCATCA-3’ GAPDH Reverse 5’-CGTGGACTGTGGTCATGAGT-3’ Supplementary material: Differentially expressed genes log2(SADS-CoV_12h/ Qvalue (SADS-CoV _12h/ Gene Symbol Control_12h) Control_12h) PTGER4 -1.03693 6.79E-04 TMEM72 -3.08132 3.66E-04 IFIT2 -1.02918 2.11E-07 FRAT2 -1.09282 4.66E-05 -
Reprogramming of Lysosomal Gene Expression by Interleukin-4 and Stat6 Brignull Et Al
Reprogramming of lysosomal gene expression by interleukin-4 and Stat6 Brignull et al. Brignull et al. BMC Genomics 2013, 14:853 http://www.biomedcentral.com/1471-2164/14/853 Brignull et al. BMC Genomics 2013, 14:853 http://www.biomedcentral.com/1471-2164/14/853 RESEARCH ARTICLE Open Access Reprogramming of lysosomal gene expression by interleukin-4 and Stat6 Louise M Brignull1†, Zsolt Czimmerer2†, Hafida Saidi1,3, Bence Daniel2, Izabel Villela4,5, Nathan W Bartlett6, Sebastian L Johnston6, Lisiane B Meira4, Laszlo Nagy2,7 and Axel Nohturfft1* Abstract Background: Lysosomes play important roles in multiple aspects of physiology, but the problem of how the transcription of lysosomal genes is coordinated remains incompletely understood. The goal of this study was to illuminate the physiological contexts in which lysosomal genes are coordinately regulated and to identify transcription factors involved in this control. Results: As transcription factors and their target genes are often co-regulated, we performed meta-analyses of array-based expression data to identify regulators whose mRNA profiles are highly correlated with those of a core set of lysosomal genes. Among the ~50 transcription factors that rank highest by this measure, 65% are involved in differentiation or development, and 22% have been implicated in interferon signaling. The most strongly correlated candidate was Stat6, a factor commonly activated by interleukin-4 (IL-4) or IL-13. Publicly available chromatin immunoprecipitation (ChIP) data from alternatively activated mouse macrophages show that lysosomal genes are overrepresented among Stat6-bound targets. Quantification of RNA from wild-type and Stat6-deficient cells indicates that Stat6 promotes the expression of over 100 lysosomal genes, including hydrolases, subunits of the vacuolar H+ ATPase and trafficking factors. -

Integrated Computational Approach to the Analysis of RNA-Seq Data Reveals New Transcriptional Regulators of Psoriasis
OPEN Experimental & Molecular Medicine (2016) 48, e268; doi:10.1038/emm.2016.97 & 2016 KSBMB. All rights reserved 2092-6413/16 www.nature.com/emm ORIGINAL ARTICLE Integrated computational approach to the analysis of RNA-seq data reveals new transcriptional regulators of psoriasis Alena Zolotarenko1, Evgeny Chekalin1, Alexandre Mesentsev1, Ludmila Kiseleva2, Elena Gribanova2, Rohini Mehta3, Ancha Baranova3,4,5,6, Tatiana V Tatarinova6,7,8, Eleonora S Piruzian1 and Sergey Bruskin1,5 Psoriasis is a common inflammatory skin disease with complex etiology and chronic progression. To provide novel insights into the regulatory molecular mechanisms of the disease, we performed RNA sequencing analysis of 14 pairs of skin samples collected from patients with psoriasis. Subsequent pathway analysis and extraction of the transcriptional regulators governing psoriasis-associated pathways was executed using a combination of the MetaCore Interactome enrichment tool and the cisExpress algorithm, followed by comparison to a set of previously described psoriasis response elements. A comparative approach allowed us to identify 42 core transcriptional regulators of the disease associated with inflammation (NFκB, IRF9, JUN, FOS, SRF), the activity of T cells in psoriatic lesions (STAT6, FOXP3, NFATC2, GATA3, TCF7, RUNX1), the hyper- proliferation and migration of keratinocytes (JUN, FOS, NFIB, TFAP2A, TFAP2C) and lipid metabolism (TFAP2, RARA, VDR). In addition to the core regulators, we identified 38 transcription factors previously not associated with the disease that can clarify the pathogenesis of psoriasis. To illustrate these findings, we analyzed the regulatory role of one of the identified transcription factors (TFs), FOXA1. Using ChIP-seq and RNA-seq data, we concluded that the atypical expression of the FOXA1 TF is an important player in the disease as it inhibits the maturation of naive T cells into the (CD4+FOXA1+CD47+CD69+PD-L1(hi) FOXP3 − ) regulatory T cell subpopulation, therefore contributing to the development of psoriatic skin lesions. -

Structural Basis of STAT2 Recognition by IRF9 Reveals Molecular Insights Into
bioRxiv preprint doi: https://doi.org/10.1101/131714; this version posted April 28, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Structural basis of STAT2 recognition by IRF9 reveals molecular insights into 2 ISGF3 function 3 Srinivasan Rengachari1, Silvia Groiss1, Juliette, Devos1, Elise Caron2, Nathalie Grandvaux2,3, 4 Daniel Panne1* 5 1European Molecular Biology Laboratory, 38042 Grenoble, France 6 2 CRCHUM - Research center, Centre Hospitalier de l'Université de Montréal, Montréal, 7 H2X0A9, Québec, Canada 8 3 Department of Biochemistry and Molecular Medicine, Université de Montréal; Faculty of 9 Medicine, Université de Montréal, Montréal, H3C 3J7, Qc, Canada 10 *Corresponding author: email– [email protected], tel: +33–476207925 11 12 13 1 bioRxiv preprint doi: https://doi.org/10.1101/131714; this version posted April 28, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 14 15 Summary 16 Cytokine signalling is mediated by the activation of distinct sets of structurally homologous JAK 17 and STAT signalling molecules, which control nuclear gene expression and cell fate. A 18 significant expansion in the gene regulatory repertoire controlled by JAK/STAT signalling has 19 arisen by the selective interaction of STATs with IRF transcription factors. -
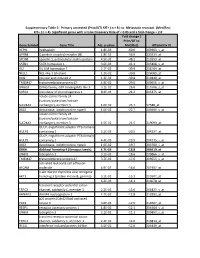
Gene Symbol Gene Title Adj. P-‐Value Fold Change ( Prim/UT to Met
Supplementary Table 2: Primary untreated (Prim/UT) KIT+ ( n = 8 ) vs. Metastatic resistant (Met/Res) KIT+ ( n = 4). Significant genes with a False Discovery Rate of < 0.05 and a fold change > 2.0 Fold change ( Prim/UT to Gene Symbol Gene Title Adj. p-value Met/Res) Affymetrix ID HEPH hephaestin 1.8E-03 -60.6 203903_s_at GPR88 G protein-coupled receptor 88 1.8E-02 -56.0 220313_at SPON1 spondin 1, extracellular matrix protein 4.5E-02 -48.2 213993_at SATB1 SATB homeobox 1 3.6E-03 -41.1 203408_s_at ISL1 ISL LIM homeobox 1 2.7E-02 -39.9 206104_at NELL1 NEL-like 1 (chicken) 1.2E-02 -39.8 206089_at RAI2 retinoic acid induced 2 1.3E-02 -30.8 219440_at TMEM47 transmembrane protein 47 4.9E-02 -29.0 209656_s_at DIRAS3 DIRAS family, GTP-binding RAS-like 3 3.1E-02 -26.8 215506_s_at SCRG1 stimulator of chondrogenesis 1 8.3E-03 -24.2 205475_at solute carrier family 24 (sodium/potassium/calcium SLC24A3 exchanger), member 3 1.6E-02 -23.7 57588_at DIO2 deiodinase, iodothyronine, type II 1.0E-02 -22.7 203699_s_at solute carrier family 24 (sodium/potassium/calcium SLC24A3 exchanger), member 3 1.5E-02 -21.5 219090_at GULP, engulfment adaptor PTB domain GULP1 containing 1 5.2E-03 -20.5 204237_at GULP, engulfment adaptor PTB domain GULP1 containing 1 4.4E-03 -19.9 204235_s_at DIO2 deiodinase, iodothyronine, type II 1.0E-02 -19.7 203700_s_at DKK4 dickkopf homolog 4 (Xenopus laevis) 4.7E-03 -18.8 206619_at LPHN3 latrophilin 3 3.1E-02 -18.6 209866_s_at TMEM47 transmembrane protein 47 3.7E-02 -17.6 209655_s_at activated leukocyte cell adhesion ALCAM molecule 4.9E-02 -
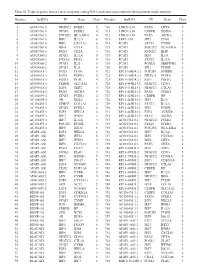
Table SI. Triple Negative Breast Cancer Long Non‑Coding RNA‑Mediated Transcriptional Dysregulation Triplet Network
Table SI. Triple negative breast cancer long non‑coding RNA‑mediated transcriptional dysregulation triplet network. Number lncRNA TF Gene Class Number lncRNA TF Gene Class 1 AC010761.8 NFATC2 FCER2 5 710 LINC01139 PAX5 CIITA 5 2 AC010761.8 STAT6 FCER2 4 711 LINC01139 CEBPB DDIT4 6 3 AC010761.8 POU2F2 HLA‑DRA 4 712 LINC01139 PAX5 AICDA 3 4 AC010761.8 IRF7 IL12A 6 713 LEF1‑AS1 SPI1 CD68 5 5 AC010761.8 IRF1 CXCL10 6 714 PCAT1 STAT1 TYMP 4 6 AC010761.8 IRF4 CIITA 5 715 PCAT1 POU2F2 HLA‑DRA 5 7 AC010761.8 PAX5 CIITA 4 716 PCAT1 RUNX2 IBSP 3 8 AC024560.2 STAT1 IL12A 5 717 PCAT1 IRF1 IL10 5 9 AC024560.2 FOXA2 PDX1 4 718 PCAT1 STAT2 IL12A 4 10 AC024560.2 STAT1 IL21 5 719 PCAT1 FOXD1 SERPINE1 3 11 AC024560.2 STAT2 IL21 5 720 PCAT1 SPI1 PTGIR 6 12 AC093818.1 PAX5 CD19 5 721 RP11‑169D4.2.1 POU2F1 GNRHR 3 13 AC093818.1 PAX5 FCER2 5 722 RP11‑169D4.2.1 NKX2‑5 PLOD1 4 14 AC093818.1 FOSL1 PLAU 4 723 RP11‑169D4.2.1 E2F1 POLE2 6 15 AC093818.1 CEBPB SLC11A1 6 724 RP11‑445K13.2 NFATC2 CD3G 5 16 AC093818.1 E2F1 TERT 3 725 RP11‑445K13.2 NFATC2 CTLA4 5 17 AC093818.1 PAX5 AICDA 5 726 RP11‑445K13.2 PAX5 FCER2 5 18 AC104699.1 STAT6 FCER2 4 727 RP11‑445K13.2 CEBPB HP 3 19 AC104699.1 AR KLK4 4 728 RP11‑445K13.2 IRF1 IL10 5 20 AC156455.1 CEBPZ COL1A1 2 729 RP11‑445K13.2 STAT1 IL12A 5 21 AC156455.1 STAT6 FCER2 4 730 RP11‑445K13.2 SPI1 PTGIR 6 22 AC156455.1 E2F1 HELLS 6 731 RP11‑445K13.2 E2F4 AURKB 6 23 AC156455.1 IRF7 IFNB1 3 732 RP11‑445K13.2 PAX5 AICDA 5 24 AC156455.1 IRF7 IL12A 3 733 AC010761.8.1 NFATC2 FCER2 5 25 AC156455.1 POU2F1 PTGIR 3 734 AC010761.8.1 STAT6 -

Loss of FOXC1 Contributes to the Corneal Epithelial Fate Switch and Pathogenesis
Signal Transduction and Targeted Therapy www.nature.com/sigtrans ARTICLE OPEN Loss of FOXC1 contributes to the corneal epithelial fate switch and pathogenesis Mingsen Li1, Liqiong Zhu1, Jiafeng Liu1, Huaxing Huang1, Huizhen Guo1, Li Wang1, Lingyu Li1, Sijie Gu1, Jieying Tan1, Jing Zhong1, Bowen Wang1, Zhen Mao1, Yong Fan2, Chunqiao Liu1, Jin Yuan1 and Hong Ouyang 1 Forkhead box C1 (FOXC1) is required for neural crest and ocular development, and mutations in FOXC1 lead to inherited Axenfeld–Rieger syndrome. Here, we find that FOXC1 and paired box 6 (PAX6) are co-expressed in the human limbus and central corneal epithelium. Deficiency of FOXC1 and alternation in epithelial features occur in patients with corneal ulcers. FOXC1 governs the fate of the corneal epithelium by directly binding to lineage-specific open promoters or enhancers marked by H3K4me2. FOXC1 depletion not only activates the keratinization pathway and reprograms corneal epithelial cells into skin-like epithelial cells, but also disrupts the collagen metabolic process and interferon signaling pathways. Loss of interferon regulatory factor 1 and PAX6 induced by FOXC1 dysfunction is linked to the corneal ulcer. Collectively, our results reveal a FOXC1-mediated regulatory network responsible for corneal epithelial homeostasis and provide a potential therapeutic target for corneal ulcer. Signal Transduction and Targeted Therapy (2021); https://doi.org/10.1038/s41392-020-00378-26:5 1234567890();,: INTRODUCTION growth of blood vessels and failure of corneal endothelial Corneal integrity and transparency are essential for normal vision. The formation.11 However, the role of FOXC1 in the corneal epithelial outermost surface of the cornea is covered with a non-keratinized homeostasis and pathogenesis has not been reported. -

Positional Specificity of Different Transcription Factor Classes Within Enhancers
Positional specificity of different transcription factor classes within enhancers Sharon R. Grossmana,b,c, Jesse Engreitza, John P. Raya, Tung H. Nguyena, Nir Hacohena,d, and Eric S. Landera,b,e,1 aBroad Institute of MIT and Harvard, Cambridge, MA 02142; bDepartment of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139; cProgram in Health Sciences and Technology, Harvard Medical School, Boston, MA 02215; dCancer Research, Massachusetts General Hospital, Boston, MA 02114; and eDepartment of Systems Biology, Harvard Medical School, Boston, MA 02215 Contributed by Eric S. Lander, June 19, 2018 (sent for review March 26, 2018; reviewed by Gioacchino Natoli and Alexander Stark) Gene expression is controlled by sequence-specific transcription type-restricted enhancers (active in <50% of the cell types) and factors (TFs), which bind to regulatory sequences in DNA. TF ubiquitous enhancers (active in >90% of the cell types) (SI Ap- binding occurs in nucleosome-depleted regions of DNA (NDRs), pendix, Fig. S1C). which generally encompass regions with lengths similar to those We next sought to infer functional TF-binding sites within the protected by nucleosomes. However, less is known about where active regulatory elements. In a recent study (5), we found that within these regions specific TFs tend to be found. Here, we char- TF binding is strongly correlated with the quantitative DNA acterize the positional bias of inferred binding sites for 103 TFs accessibility of a region. Furthermore, the TF motifs associated within ∼500,000 NDRs across 47 cell types. We find that distinct with enhancer activity in reporter assays in a cell type corre- classes of TFs display different binding preferences: Some tend to sponded closely to those that are most enriched in the genomic have binding sites toward the edges, some toward the center, and sequences of active regulatory elements in that cell type (5). -

Quantitative Profiling of BATF Family Proteins/JUNB/IRF Hetero-Trimers
Chang et al. BMC Molecular Biol (2018) 19:5 https://doi.org/10.1186/s12867-018-0106-7 BMC Molecular Biology RESEARCH ARTICLE Open Access Quantitative profling of BATF family proteins/JUNB/IRF hetero‑trimers using Spec‑seq Yiming K. Chang, Zheng Zuo and Gary D. Stormo* Abstract Background: BATF family transcription factors (BATF, BATF2 and BATF3) form hetero-trimers with JUNB and either IRF4 or IRF8 to regulate cell fate in T cells and dendritic cells in vivo. While each combination of the hetero-trimer has a distinct role, some degree of cross-compensation was observed. The basis for the diferential actions of IRF4 and IRF8 with BATF factors and JUNB is still unknown. We propose that the diferences in function between these hetero-trimers may be caused by diferences in their DNA binding preferences. While all three BATF family transcrip- tion factors have similar binding preferences when binding as a hetero-dimer with JUNB, the cooperative binding of IRF4 or IRF8 to the hetero-dimer/DNA complex could change the preferences. We used Spec-seq, which allows for the efcient and accurate determination of relative afnity to a large collection of sequences in parallel, to fnd diferences between cooperative DNA binding of IRF4, IRF8 and BATF family members. Results: We found that without IRF binding, all three hetero-dimer pairs exhibit nearly the same binding preferences to both expected wildtype binding sites TRE (TGA(C/G)TCA) and CRE (TGACGTCA). IRF4 and IRF8 show the very similar DNA binding preferences when binding with any of the three hetero-dimers. -

The Effects of Disease Mutations on the Transcription Factor Interferon Regulatory Factor 6 June 2011
The effects of disease mutations on the transcription factor Interferon Regulatory Factor 6 A thesis to the University of Manchester for the degree of Doctor of Philosophy in the Faculty of Life Science 2011 Ling-I Su - 0 - Contents Contents ........................................................................................................................... 1 List of Figures .................................................................................................................. 4 List of Tables ................................................................................................................... 7 List of Abbreviations....................................................................................................... 8 Abstract .......................................................................................................................... 12 Declaration ..................................................................................................................... 13 Copyright statement...................................................................................................... 13 Acknowledgements ........................................................................................................ 14 Chapter 1 ................................................................................................ 15 Introduction ................................................................................................................... 15 1. Introduction ..............................................................................................................