KLEIN-DISSERTATION-2014.Pdf (5.178Mb)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Identification of Transcriptional Mechanisms Downstream of Nf1 Gene Defeciency in Malignant Peripheral Nerve Sheath Tumors Daochun Sun Wayne State University
Wayne State University DigitalCommons@WayneState Wayne State University Dissertations 1-1-2012 Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors Daochun Sun Wayne State University, Follow this and additional works at: http://digitalcommons.wayne.edu/oa_dissertations Recommended Citation Sun, Daochun, "Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors" (2012). Wayne State University Dissertations. Paper 558. This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. IDENTIFICATION OF TRANSCRIPTIONAL MECHANISMS DOWNSTREAM OF NF1 GENE DEFECIENCY IN MALIGNANT PERIPHERAL NERVE SHEATH TUMORS by DAOCHUN SUN DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2012 MAJOR: MOLECULAR BIOLOGY AND GENETICS Approved by: _______________________________________ Advisor Date _______________________________________ _______________________________________ _______________________________________ © COPYRIGHT BY DAOCHUN SUN 2012 All Rights Reserved DEDICATION This work is dedicated to my parents and my wife Ze Zheng for their continuous support and understanding during the years of my education. I could not achieve my goal without them. ii ACKNOWLEDGMENTS I would like to express tremendous appreciation to my mentor, Dr. Michael Tainsky. His guidance and encouragement throughout this project made this dissertation come true. I would also like to thank my committee members, Dr. Raymond Mattingly and Dr. John Reiners Jr. for their sustained attention to this project during the monthly NF1 group meetings and committee meetings, Dr. -

Goyette Guillaume 2009 These.Pdf (8.250Mb)
Université de Montréal Caractérisation moléculaire de la modulation spatio- temporelle des fonctions du phagosome Par Guillaume Goyette Département de pathologie et biologie cellulaire Faculté de médecine Thèse présentée à la faculté des études supérieures En vue de l’obtention du grade Philosophiae Doctor (Ph.D.) en pathologie et biologie cellulaire 24 avril 2009 © Guillaume Goyette 2009 ii Université de Montréal Faculté des études supérieures Cette thèse intitulée : Caractérisation moléculaire de la modulation spatio-temporelle des fonctions du phagosome présentée par : Guillaume Goyette a été évaluée par un jury composé des personnes suivantes : Jacques Paiement, président-rapporteur Michel Desjardins, directeur de recherche Dorin-Lucian Ghitescu, membre du jury John J.M. Bergeron, examinateur externe Nicole Leclerc, représentante du doyen de la FES iii Résumé La phagocytose est un processus par lequel des cellules spécialisées du système immunitaire comme les macrophages ingèrent des microorganismes envahisseurs afin de les détruire. Les microbes phagocytés se retrouvent dans un compartiment intracellulaire nommé le phagosome, qui acquiert graduellement de nombreuses molécules lui permettant de se transformer en phagolysosome possédant la capacité de tuer et dégrader son contenu. L’utilisation de la protéomique a permis de mettre en évidence la présence de microdomaines (aussi nommés radeaux lipidiques ou radeaux membranaires) sur les phagosomes des macrophages. Notre équipe a démontré que ces radeaux exercent des fonctions cruciales au niveau de la membrane du phagosome. D’abord nous avons observé que la survie du parasite intracellulaire L. donovani est possible dans un phagosome dépourvu de radeaux lipidiques. Parallèlement nous avons constaté qu’un mutant de L. donovani n’exprimant pas de LPG à sa surface (LPG-) est rapidement tué dans un phagosome arborant des radeaux membranaires. -

University of São Paulo
UNIVERSITY OF SÃO PAULO PHARMACEUTICAL SCIENCES FACULTY Department of Clinical and Toxicological Analysis Graduate Program in Pharmacy (Physiopathology and Toxicology) A non-targeted proteomics investigation of cylindrospermopsin-induced hepatotoxicity Carlos Andrey González Blanco Manuscript presented for the degree of P.h.D. Professor Advisor: Assoc. Prof. Ernani Pinto São Paulo 2017 UNIVERSIDADE DE SÃO PAULO FACULDADE DE CIÊNCIAS FARMACÊUTICAS Departamento de Analises Clinicas e Toxicológicas Programa de Pós-graduação em Farmacia (Fisiopatologia e Toxicologia) Uso de uma estratégia proteômica não-direcionada para investigar a hepatotoxicidade producida pela cilindrospermopsina Carlos Andrey González Blanco Versão corrigida da Tese conforme resolução CoPGr 6018 Tese para obtenção do Título de Doutor Orientador: Prof. Assoc. Ernani Pinto São Paulo 2017 CARLOS ANDREY GONZÁLEZ BLANCO A non-targeted proteomics investigation of cylindrospermopsin-induced hepatotoxicity Comissão Julgadora da Tese para obtenção do Título de Doutor Prof. Assoc. Ernani Pinto orientador/presidente _________________________________________________ 1o. examinador _________________________________________________ 2o. examinador __________________________________________________ 3o. examinador __________________________________________________ 4o. examinador São Paulo, ______ de _______________ de 2017 i “The map is not the territory”. A. Korzybski ii Acknowledgement I would like to thank my thesis advisor Prof. Ernani for his unconditional support and friendship. Throughout the years, I saw you taking risks in projects and making daring decisions in your academic career, which always turned up right. The audacity I saw in your work inspired me to strive harder and try out new ideas. I would also like to thank all the students I met throught the years at prof. Ernani`s lab, all of which are and will always be great friends; I learned something from each and every one of you. -

How Abnormal RNA Metabolism Results in Childhood-Onset Neurological Diseases
Exosomal Protein Deficiencies: How Abnormal RNA Metabolism Results in Childhood-Onset Neurological Diseases A thesis submitted for the degree of Doctor of Philosophy at Newcastle University October 2016 Michele Giunta Institute of Genetic Medicine ii Author’s declaration This thesis is submitted for the degree of Doctor of Philosophy at Newcastle University. I, Michele Giunta, declare that the work described here is my own, unless where clearly acknowledged and stated otherwise. I certify that I have not submitted any of the material in this thesis for a degree qualification at this or any other university. iii Abstract RNA metabolism is of critical importance for normal cellular functions and needs to be finely tuned in order to maintain stable conditions within the cell. The exosome complex is the most important RNA processing machinery, responsible for the correct processing of many different types of RNAs and interacting with different co-factors which bind and carry specific subtypes of RNA for degradation to the complex. Mutations in exosome complex subunits (EXOSC3, EXOSC8) were reported to cause severe childhood onset complex neurological disorders presenting with pontocerebellar hypoplasia type 1 (PCH1), spinal muscular atrophy (SMA) and central nervous system hypomyelination. We have recently identified a homozygous pathogenic mutation in RNA Binding Motif Protein 7 RBM7, a subunit of the nuclear exosome targeting (NEXT) complex in a single patient with SMA-like phenotype and proved that RBM7 is a novel human disease gene related to the exosome complex. In order to understand the disease mechanism in RBM7 deficiency and to explore the role of exosome complex in neurodevelopment, we performed gene expression studies (RT-PCR, RNA sequencing) in human cells of patients carrying mutations in EXOSC8 and RBM7. -

Mrna Expression in Human Leiomyoma and Eker Rats As Measured by Microarray Analysis
Table 3S: mRNA Expression in Human Leiomyoma and Eker Rats as Measured by Microarray Analysis Human_avg Rat_avg_ PENG_ Entrez. Human_ log2_ log2_ RAPAMYCIN Gene.Symbol Gene.ID Gene Description avg_tstat Human_FDR foldChange Rat_avg_tstat Rat_FDR foldChange _DN A1BG 1 alpha-1-B glycoprotein 4.982 9.52E-05 0.68 -0.8346 0.4639 -0.38 A1CF 29974 APOBEC1 complementation factor -0.08024 0.9541 -0.02 0.9141 0.421 0.10 A2BP1 54715 ataxin 2-binding protein 1 2.811 0.01093 0.65 0.07114 0.954 -0.01 A2LD1 87769 AIG2-like domain 1 -0.3033 0.8056 -0.09 -3.365 0.005704 -0.42 A2M 2 alpha-2-macroglobulin -0.8113 0.4691 -0.03 6.02 0 1.75 A4GALT 53947 alpha 1,4-galactosyltransferase 0.4383 0.7128 0.11 6.304 0 2.30 AACS 65985 acetoacetyl-CoA synthetase 0.3595 0.7664 0.03 3.534 0.00388 0.38 AADAC 13 arylacetamide deacetylase (esterase) 0.569 0.6216 0.16 0.005588 0.9968 0.00 AADAT 51166 aminoadipate aminotransferase -0.9577 0.3876 -0.11 0.8123 0.4752 0.24 AAK1 22848 AP2 associated kinase 1 -1.261 0.2505 -0.25 0.8232 0.4689 0.12 AAMP 14 angio-associated, migratory cell protein 0.873 0.4351 0.07 1.656 0.1476 0.06 AANAT 15 arylalkylamine N-acetyltransferase -0.3998 0.7394 -0.08 0.8486 0.456 0.18 AARS 16 alanyl-tRNA synthetase 5.517 0 0.34 8.616 0 0.69 AARS2 57505 alanyl-tRNA synthetase 2, mitochondrial (putative) 1.701 0.1158 0.35 0.5011 0.6622 0.07 AARSD1 80755 alanyl-tRNA synthetase domain containing 1 4.403 9.52E-05 0.52 1.279 0.2609 0.13 AASDH 132949 aminoadipate-semialdehyde dehydrogenase -0.8921 0.4247 -0.12 -2.564 0.02993 -0.32 AASDHPPT 60496 aminoadipate-semialdehyde -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses by a Synthetic TLR3 Ligand Mitigates
Downloaded from http://www.jimmunol.org/ by guest on September 28, 2021 Inducing is online at: average * The Journal of Immunology published online 16 August 2013 from submission to initial decision 4 weeks from acceptance to publication http://www.jimmunol.org/content/early/2013/08/16/jimmun ol.1300376 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol Submit online. Every submission reviewed by practicing scientists ? is published twice each month by http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 28, 2021. Published August 16, 2013, doi:10.4049/jimmunol.1300376 The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

1 a Novel Prognostic Two-Gene Signature for Triple Negative Breast
A Novel Prognostic Two-Gene Signature for Triple Negative Breast Cancer Mansour A Alsaleem 1,2, Graham Ball 3, Michael S Toss 1, Sara Raafat 1, Mohammed Aleskandarany 1,4, Chitra Joseph 1, Angela Ogden 5, Shristi Bhattarai 5, Padmashree C G Rida 5, Francesca Khani 6, Melissa Davis 7, Olivier Elemento 8, Ritu Aneja 5, Ian O Ellis 1, Andrew Green 1, Nigel P Mongan 9,10 and Emad Rakha 1,4,11. 1 Nottingham Breast Cancer Research Centre, Division of Cancer and Stem Cells, School of Medicine, University of Nottingham, Nottingham, UK. 2 Faculty of Applied Medical Sciences, Onizah Community College, Qassim University, Qassim, Saudi Arabia. 3 John van Geest Cancer Research Centre, Nottingham Trent University, Nottingham, UK 4 Faculty of Medicine, Menoufyia University, Shebin El Kom, Egypt. 5 Department of Biology, Georgia State University, Atlanta, GA, USA. 6 Department of Pathology and Laboratory Medicine, Weill Cornell Medical College, New York, NY, USA 7 Department of Genetics, Franklin College of Arts and Sciences, University of Georgia, Athens, GA, USA. 8 Institute for Computational Biomedicine, Department of Physiology and Biophysics, Weill Cornell Medicine of Cornell University, New York, NY, USA 9 University of Nottingham Biodiscovery Institute, Faculty of Medicine and Health Sciences, School of Veterinary Medicine and Science, University of Nottingham, Nottingham, UK. 10 Department of Pharmacology, Weill Cornell Medicine, New York, NY, USA. 11 Department of Histopathology, Division of Cancer and Stem Cells, School of Medicine, The University -

Mechanistic Insight Into WEB-2170-Induced Apoptosis In
CORE Metadata, citation and similar papers at core.ac.uk Provided by Florence Research Experimental Hematology 2009;37:1176–1185 Mechanistic insight into WEB-2170-induced apoptosis in human acute myelogenous leukemia cells: The crucial role of PTEN Cristina Cellaia,*, Anna Laurenzanaa,*, Elisa Bianchib, Sara Sdelcia, Rossella Manfredinib, Alessandro M. Vannucchic, Roberto Caporalec, Manjola Balliua, Francesco Mannellic, Sergio Ferrarib, Alberto Bosib, Debora Miniatid, Pier L. Coccoa, Steeve Veronneaue, Jana Stankovae, and Francesco Paolettia aDepartment of Experimental Pathology and Oncology, University of Florence, Florence, Italy; bDepartment of Biological Sciences, University of Modena e Reggio Emilia, Modena, Italy; cDepartment of Hematology, University of Florence, Florence, Italy; dDepartment of Preclinical and Clinical Pharmacology, University of Florence, Florence, Italy; eDepartment of Pediatrics, Universite`de Sherbrooke, Sherbrooke, Que´bec, Canada (Received 9 April 2009; revised 29 June 2009; accepted 9 July 2009) Objective. This study aimed to investigate the mechanisms of action of WEB-2170, an inverse agonist of platelet-activating factor receptor, capable of inducing apoptosis in human acute myelogenous leukemia (AML) cells. Material and Methods. Gene expression profiling followed by cytofluorimetric, morphologic, and biologic analyses were used to monitor WEB-2170 effects in AML cell lines (ie, NB4, KG1, NB4-MR4, THP1, and U937) and blasts from patients with different AML (M0LM5) subtypes. PTEN silencing with small interfering -
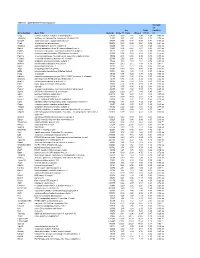
Table S-1 Mpkccd Transcriptome
Table S1. mpkCCD Cell Transcriptomes. Correlati on Ori. Ratio Coefficie Gene Symbol Gene Title GeneID Clone 11 Clone Clone 2 (11/2) nt Copg coatomer protein complex, subunit gamma 54161 1.99 1.81 2.48 0.80 -0.49 ns Atp6v0d1 ATPase, H+ transporting, lysosomal V0 subunit D1 11972 3.37 2.87 3.00 1.12 0.74 ns Golga7 golgi autoantigen, golgin subfamily a, 7 57437 3.03 3.40 3.58 0.84 -0.34 ns Psph phosphoserine phosphatase 100678 0.38 0.66 0.64 0.59 -0.61 ns Trappc4 trafficking protein particle complex 4 60409 1.17 1.12 1.08 1.08 0.64 ns Dpm2 dolichol-phosphate (beta-D) mannosyltransferase 2 13481 0.74 0.87 0.81 0.91 -0.31 ns Psmb5 proteasome (prosome, macropain) subunit, beta type 5 19173 2.92 3.00 2.92 0.99 0.32 ns Dhrs1 dehydrogenase/reductase (SDR family) member 1 52585 0.74 0.68 0.88 0.83 -0.18 ns Ppm1a protein phosphatase 1A, magnesium dependent, alpha isoform 19042 2.43 2.60 2.96 0.82 -0.53 ns Psenen presenilin enhancer 2 homolog (C. elegans) 66340 2.52 2.88 3.80 0.66 -0.77 ns Anapc1 anaphase promoting complex subunit 1 17222 1.16 1.33 1.61 0.72 -0.33 ns Mrpl43 mitochondrial ribosomal protein L43 94067 2.15 2.15 1.84 1.16 0.91 * Nmt1 N-myristoyltransferase 1 18107 3.21 2.71 3.36 0.95 0.31 ns Atg5 autophagy-related 5 (yeast) 11793 0.70 0.73 0.66 1.05 -0.23 ns Mtif2 mitochondrial translational initiation factor 2 76784 1.16 1.29 1.19 0.97 -0.36 ns Psap prosaposin 19156 8.07 7.25 8.37 0.96 0.02 ns Ube2g1 ubiquitin-conjugating enzyme E2G 1 (UBC7 homolog, C. -

Weighted Gene Co-Expression Network Analysis and Machine Learning Identify Critical Genes in the Development of Osteomyelitis After Staphylococcus Aureus Infections
Weighted Gene Co-Expression Network Analysis and Machine learning Identify Critical Genes in the Development of Osteomyelitis after Staphylococcus aureus Infections Baixing Chen KU Leuven Shaoshuo Li Nanjing University of Chinese Medicine Zhaoqi Lu The Fourth Clinical Medical College, Guangzhou University of Chinese Medicine,China Mingling Huang Guangzhou University of Chinese Medicine Shi Lin Guangzhou University of Chinese Medicine Hang Dong ( [email protected] ) The First Aliated Hospital of Guangzhou University of Chinese Medicine Research Article Keywords: Osteomyelitis, bone infections, WGCNA, machine learning, random forest, development, Staphylococcus aureus, GEO, recursive feature elimination, microarray Posted Date: April 27th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-440346/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract Background: Staphylococcus aureus (S. aureus) is the most common pathogen that causes osteomyelitis (OM). However, OM's pathogenesis, which is not clear, involves many factors such as environment, genetics and immunity dysregulation. This study aims to explore the key genes involved in the pathogenesis and development of OM following S. aureus infection. Methods: After obtaining the datasets of GSE6269 and GSE16129, we performed weighted gene co- expression network analysis (WGCNA) to nd clusters modules of highly correlated genes and recursive feature elimination (RFE) method to narrow the range of feature genes. For determining the effect of feature genes, we constructed a random forest (RF) model with feature genes and validated the predictive validity of the RF model using independent data from GSE11908. The protein-protein interaction (PPI) network identies essential proteins that contributed to OM development. -
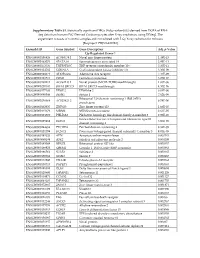
Supplementary Table S1. Statistically Significant Degs (Adj.P
Supplementary Table S1. Statistically significant DEGs (Adj.p-value<0.05) derived from DGEA of RNA- Seq data from human iPSC-Derived Cardiomyocytes after X-ray irradiation, using DESeq2. The experiment consists of 3 control samples and 3 irradiated with 5 Gy X-ray radiation for 48 hours [Bioproject: PRJNA421022]. Ensembl ID Gene Symbol Gene Description Adj. p-Value Up-Regulated Genes↑ ENSG00000196826 AC008758.1 Novel zinc finger protein 1.48E-13 ENSG00000163071 SPATA18 Spermatogenesis associated 18 2.89E-13 ENSG00000173535 TNFRSF10C TNF receptor superfamily member 10c 2.50E-11 ENSG00000124762 CDKN1A Cyclin dependent kinase inhibitor 1A 3.30E-09 ENSG00000128271 ADORA2A Adenosine A2a receptor 7.41E-09 ENSG00000161513 FDXR Ferredoxin reductase 3.25E-07 ENSG00000198211 AC092143.1 Novel protein (MC1R-TUBB3 readthrough) 1.03E-06 ENSG00000270181 BIVM-ERCC5 BIVM-ERCC5 readthrough 4.30E-06 ENSG00000177106 EPS8L2 EPS8 like 2 6.65E-06 ENSG00000080546 SESN1 Sestrin 1 7.62E-06 Ribosomal L24 domain containing 1 (RSL24D1) ENSG00000256664 AC025423.2 9.78E-06 pseudogene ENSG00000102935 ZNF423 Zinc finger protein 423 1.66E-05 ENSG00000135679 MDM2 MDM2 proto-oncogene 2.63E-05 ENSG00000174307 PHLDA3 Pleckstrin homology like domain family A member 3 2.92E-05 Extracellular leucine rich repeat and fibronectin type III ENSG00000225968 ELFN1 3.06E-05 domain containing 1 ENSG00000244694 PTCHD4 Patched domain containing 4 8.94E-05 ENSG00000131398 KCNC3 Potassium voltage-gated channel subfamily C member 3 9.03E-05 ENSG00000181026 AEN Apoptosis enhancing nuclease