The Nervous System – Target Organ Into the Twenty-First Century
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Carbamate Pesticides Aldicarb Aldicarb Sulfoxide Aldicarb Sulfone
Connecticut General Statutes Sec 19a-29a requires the Commissioner of Public Health to annually publish a list setting forth all analytes and matrices for which certification for testing is required. Connecticut ELCP Drinking Water Analytes Revised 05/31/2018 Microbiology Total Coliforms Fecal Coliforms/ E. Coli Carbamate Pesticides Legionella Aldicarb Cryptosporidium Aldicarb Sulfoxide Giardia Aldicarb Sulfone Carbaryl Physicals Carbofuran Turbidity 3-Hydroxycarbofuran pH Methomyl Conductivity Oxamyl (Vydate) Minerals Chlorinated Herbicides Alkalinity, as CaCO3 2,4-D Bromide Dalapon Chloride Dicamba Chlorine, free residual Dinoseb Chlorine, total residual Endothall Fluoride Picloram Hardness, Calcium as Pentachlorophenol CaCO3 Hardness, Total as CaCO3 Silica Chlorinated Pesticides/PCB's Sulfate Aldrin Chlordane (Technical) Nutrients Dieldrin Endrin Ammonia Heptachlor Nitrate Heptachlor Epoxide Nitrite Lindane (gamma-BHC) o-Phosphate Metolachlor Total Phosphorus Methoxychlor PCB's (individual aroclors) Note 1 PCB's (as decachlorobiphenyl) Note 1 Demands Toxaphene TOC Nitrogen-Phosphorus Compounds Alachlor Metals Atrazine Aluminum Butachlor Antimony Diquat Arsenic Glyphosate Barium Metribuzin Beryllium Paraquat Boron Propachlor Cadmium Simazine Calcium Chromium Copper SVOC's Iron Benzo(a)pyrene Lead bis-(2-ethylhexyl)phthalate Magnesium bis-(ethylhexyl)adipate Manganese Hexachlorobenzene Mercury Hexachlorocyclopentadiene Molybdenum Nickel Potassium Miscellaneous Organics Selenium Dibromochloropropane (DBCP) Silver Ethylene Dibromide (EDB) -

Four Patients with Amanita Phalloides Poisoning CASE SERIE
CASE SERIE 353 Four patients with Amanita Phalloides poisoning S. Vanooteghem1, J. Arts2, S. Decock2, P. Pieraerts3, W. Meersseman4, C. Verslype1, Ph. Van Hootegem2 (1) Department of hepatology, UZ Leuven ; (2) Department of gastroenterology, AZ St-Lucas, Brugge ; (3) General Practitioner, Zedelgem ; (4) Department of internal medicine, UZ Leuven. Abstract because they developed stage 2 hepatic encephalopathy. With maximal supportive therapy, all patients gradually Mushroom poisoning by Amanita phalloides is a rare but poten- improved from day 3 and recovered without the need for tially fatal disease. The initial symptoms of nausea, vomiting, ab- dominal pain and diarrhea, which are typical for the intoxication, liver transplantation. They were discharged from the can be interpreted as a common gastro-enteritis. The intoxication hospital between 6 to 10 days after admission. can progress to acute liver and renal failure and eventually death. Recognizing the clinical syndrome is extremely important. In this case report, 4 patients with amatoxin intoxication who showed the Discussion typical clinical syndrome are described. The current therapy of amatoxin intoxication is based on small case series, and no ran- Among mushroom intoxications, amatoxin intoxica- domised controlled trials are available. The therapy of amatoxin intoxication consists of supportive care and medical therapy with tion accounts for 90% of all fatalities. Amatoxin poison- silibinin and N-acetylcysteine. Patients who develop acute liver fail- ing is caused by mushroom species belonging to the gen- ure should be considered for liver transplantation. (Acta gastro- era Amanita, Galerina and Lepiota. Amanita phalloides, enterol. belg., 2014, 77, 353-356). commonly known as the “death cap”, causes the majority Key words : amanita phalloides, mushroom poisoning, acute liver of fatal cases. -

Clinical Biochemistry of Hepatotoxicity
linica f C l To o x l ic a o n r l o u g Singh, J Clinic Toxicol 2011, S:4 o y J Journal of Clinical Toxicology DOI: 10.4172/2161-0495.S4-001 ISSN: 2161-0495 ReviewResearch Article Article OpenOpen Access Access Clinical Biochemistry of Hepatotoxicity Anita Singh1, Tej K Bhat2 and Om P Sharma2* 1CSK Himachal Pradesh, Krishi Vishva Vidyalaya, Palampur (HP) 176 062, India 2Biochemistry Laboratory, Indian Veterinary Research Institute, Regional Station, Palampur (HP) 176 061, India Abstract Liver plays a central role in the metabolism and excretion of xenobiotics which makes it highly susceptible to their adverse and toxic effects. Liver injury caused by various toxic chemicals or their reactive metabolites [hepatotoxicants) is known as hepatotoxicity. The present review describes the biotransformation of hepatotoxicants and various models used to study hepatotoxicity. It provides an overview of pathological and biochemical mechanism involved during hepatotoxicity together with alteration of clinical biochemistry during liver injury. The review has been supported by a list of important hepatotoxicants as well as common hepatoprotective herbs. Keywords: Hepatotoxicity; Hepatotoxicant; In Vivo models; In Vitro production of bile thus leading to the body’s inability to flush out the models; Pathology; Alanine aminotransferase; Alkaline phosphatase; chemicals through waste. Smooth endoplasmic reticulum of the liver is Bilirubin; Hepatoprotective the principal ‘metabolic clearing house’ for both endogenous chemicals like cholesterol, steroid hormones, fatty acids and proteins, and Introduction exogenous substances like drugs and alcohol. The central role played by liver in the clearance and transformation of chemicals exposes it to Hepatotoxicity refers to liver dysfunction or liver damage that is toxic injury [4]. -

Impact of Pesticide Use on Health in Developing Countries
Impact of pesticide use on health in developing countries Proceedings of a symposium held in Ottawa, Canada, 1 7-20 September 1990 IDRC CRDI International Development Research Centre Centre de recherches pour le devetoppement international 1 March 1993 Dear Reader/Librarian, IDRC is a public corporation created by the Canadian parliament in 1970 to help developing countries find viable solutions to their problems through research. At the 1992 Earth Summit, IDRC's mandate was broadened to emphasize sustainable development issues. As part of IDRC's strengthened commitment to global action and harüony, we are pleased to send you a complimentary copy of our most recent publication: The impact of pesticide use on health in developing countries (March 1993, 352 pages, 0-88936-560-1, $17.95). The first part of this book presents a brief survey of the global situation and the results of twelve epidemiological studies carried out by researchers from Africa, Latin America, Asia and the Middle East. These focus on poisonings resulting from organophosphates, herbicides, and pyrethroids. The second part illustrates the role of the process of development, production, spraying techniques and legislation in protecting the health of workers. A discussion of the benefits and modalities of access to pertinent information for the prevention of pesticide poisonings is provided in the third section. Finally, in the fourth section, consideration is given to the advantages and disadvantages of certain alternatives to the use of synthetic pesticides in agriculture and public health, such as botanical pesticides and integrated pest management strategies. We hope this book is a valuable addition to your collection. -

Teratogenicity and Embryotoxicity of Organophosphorus Compounds in Animal Models - a Short Review
Mil. Med. Sci. Lett. (Voj. Zdrav. Listy) 2012, vol. 81(1), p. 16-26 ISSN 0372-7025 DOI: 10.31482/mmsl.2012.003 REVIEW ARTICLE TERATOGENICITY AND EMBRYOTOXICITY OF ORGANOPHOSPHORUS COMPOUNDS IN ANIMAL MODELS - A SHORT REVIEW Syed M Nurulain and M Shafiullah Department of Pharmacology and Therapeutic, Faculty of Medicine and Health Sciences, UAE University, AlAin, UAE. P.O.Box 17666, Alain, UAE Received 9 th December 2011. Revised 12 th February 2012. Published 9 th March 2012. Summary Organophosphorus compounds (OPCs) are a wide group of compounds both structurally and functionally. Each OPC has a unique toxicological profile. The exposure to this type of poison is not limited only to certain occupationally exposed people but also to children, women, pregnant women; all have chances to be exposed to this poison. During the recent past years it has been reported in many poison epidemiological studies and case reports that exposure of OPCs during pregnancy caused malformed fetuses, neural tube defect (NTD) and shortening of pregnancy. The literature for animal models reveals inconclusive evidence. The generalized view is that they are neither teratogenic nor embryotoxic. But it is not true. There is a lack of systematic study and scarcity of reports on the topic. The present study was undertaken to investigate the teratogenicity induced by organophosphorus compounds in different animal models by literature review. Literature was searched by Toxicology Data NetWork (TOXNET), Developmental and Reproductive Toxicology Database (DART), Toxicology Literature Online (TOXLINE), Hazardous Substances Data Bank (HSDB), Pubmed Central, Entrez-Pubmed, Science Direct, Directory Of Open Access Journal (DOAJ), Google Scholar and International Program on Chemical Safety (IPCS-INCHEM), Embase. -

Recreational Noise-Induced Hearing Loss
Hearing loss due to recreational exposure to loud sounds A review World Health Organization Hearing loss due to recreational exposure to loud sounds A review World Health Organization Contributors: Etienne Krug, Maria Alarcos Cieza, Shelly Chadha, Laura Sminkey, Thais Morata, DeWet Swanepoel, Adrian Fuente, Warwick Williams, Joseph Cerquone, Ricardo Martinez, Gretchen Stevens, Margie Peden, Sowmya Rao, Paras Agarwal, Eighmey Zeeck, Anna Bladey, Malachi Arunda, Aileen Ncube. Graphics Credits: INIS Communications WHO Library Cataloguing-in-Publication Data Hearing loss due to recreational exposure to loud sounds: a review. 1.Hearing Loss, Noise-Induced. 2.Music. 3.Noise. 4.Recreation. 5.Noise. Transportation. 6.Adolescent. I.World Health Organization. ISBN 978 92 4 150851 3 (NLM classification: WV 270) © World Health Organization 2015 All rights reserved. Publications of the World Health Organization are available on the WHO website (http://www.who.int) or can be purchased from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for non- commercial distribution – should be addressed to WHO Press through the WHO website (http://www.who.int/about/licensing/copyright_form/en/index.html). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. -

Organophosphate Poisoning : a Review
120 Sinha and Sharma Med J Indones Organophosphate poisoning : A review Parmod K. Sinha, Ashok Sharma Abstrak Pestisida organofosfat digunakan secara luas di seluruh dunia. Keracunan oleh bahan ini merupakan masalah kesehatan masyarakat, terutama di negara berkembang. Zat neurotoksik organofosfat merupakan bahan yang dianggap mengancam dalam bidang militer dan terorisme. Mekanisme toksisitas bahan ini adalah dengan cara menghambat asetilkolinesterase yang mengakibatkan menumpuknya neurotransmitor asetilkolin dan terjadi rangsangan terus-menerus pada reseptor asetilkolin pada sistem saraf sentral maupun perifer. Selain krisis kolinergik, organofosfat dapat menimbulkan berbagai sindrom neurologis, baik akut maupun kronik. Sedangkan gejala peralihan ( intermediate) terjadi 1-4 hari setelah krisis kolinergik teratasi. Pengobatan standar terdiri dari reaktivasi asetilkolinesterase dengan antidot golongan oksim (prolidoksim, oksidoksime, HI-6 dan HLo7), dan pengendalian efek biokimia asetilkolin dengan menggunakan atropin. Golongan oksim yang baru HI-6 dan Hlo7 merupakan reaktivator asetilkolinesterase yang lebih cocok dan efektif untuk keracunan akut dan berat dibandingkan dengan prolidoksim dan obidoksim. Penderita yang mendapat pengobatan segera, biasanya dapat sembuh dari toksisitas akut, namun gejala neurologis ikutan dapat saja terjadi. (Med J Indones 2003; 12: 120-6) Abstract Organophosphate pesticides are used extensively worldwide, and poisoning by these agents, particularly in developing nations is a public health problem. Organophosphorous -

Guide No. 1 – October 2020 2/12 the CONCEPT and IMPLEMENTATION of CPA GUIDANCE RESIDUE LEVELS
Cooperation Centre for Scientific Research Relative to Tobacco CORESTA GUIDE N° 1 The Concept and Implementation of CPA Guidance Residue Levels October 2020 Agro-Chemical Advisory Committee CORESTA TECHNICAL GUIDE N° 1 Title: The Concept and Implementation of CPA Guidance Residue Levels Status: Valid Note: This document will be periodically reviewed by CORESTA Document history: Date of review Information July 2003 Version 1 GRL for Pyrethrins () and Terbufos corrected. December 2003 CPA terminology corrected. June 2008 Version 2 – GRLs revised and residue definitions added Provisional GRL of 2.00 ppm for Cyfluthrin to replace previous June 2010 GRL of 0.50 ppm July 2013 Version 3 – GRLs revised October 2013 Note for Maleic Hydrazide revised Version 4 – GRLs revised + clarification that scope of GRLs July 2016 applies predominantly to the production of traditional cigarette tobaccos and GAP associated with their cultivation. June 2018 Fluopyram GRL of 5 ppm added to GRL list Version 5 – Nine new CPAs with GRL added to list. November 2019 Revision of GRLs for Chlorantraniliprole and Indoxacarb. Updated web links. October 2020 Version 6 – Flupyradifurone GRL of 21 ppm added to GRL list. CORESTA Guide No. 1 – October 2020 2/12 THE CONCEPT AND IMPLEMENTATION OF CPA GUIDANCE RESIDUE LEVELS Executive Summary • Guidance Residue Levels (GRLs) are in the remit of the Agro-Chemical Advisory Committee (ACAC) of CORESTA. Their development is a joint activity of all ACAC members, who represent the leaf production, processing and manufacturing sectors of the Tobacco Industry. The concept of GRLs and their implementation are described in this guide. • GRLs provide guidance to tobacco growers and assist with interpretation and evaluation of results from analyses of residues of Crop Protection Agents (CPAs*). -

P-Listed Hazardous Wastes
P-Listed Hazardous Wastes The Environmental Protection Agency (EPA) has identified a number of chemicals on the EPA “P-list” that present an especially acute hazard when disposed of as hazardous waste. Because of their acute hazards, there are more stringent requirements when disposing of these wastes: ►Container size: When collecting p-listed chemicals as waste, the volume of the hazardous waste container must not exceed one quart (approximately one liter). ►Empty containers: Empty containers that held p-listed chemicals must also be disposed of as hazardous waste. They are not allowed to be washed or re-used. ►Contaminated materials: Disposable materials that become contaminated with p-listed chemicals (e.g. gloves, weighing boats, etc.) must also be disposed of as hazardous waste. Non-disposable materials must be “triple-rinsed”, or rinsed three times to remove the contamination. This rinsate must be collected as hazardous waste. Materials contaminated with p-listed chemicals may not be washed or re-used until they have been triple-rinsed. Remember: - Label the waste as hazardous waste. Most common p-listed wastes Just like all other hazardous wastes, p-listed Chemical CAS number wastes must be labeled with the words Acrolein 107–02–8 “hazardous waste”, the complete chemical Allyl alcohol 107–18–6 name, and the associated hazard Arsenic compounds Varies characteristics (e.g., ignitable, corrosive, Inorganic cyanide Varies toxic, or reactive). salts Carbon disulfide 75-15-0 - Use disposable materials whenever Cyanogen and 460-19-5, 506-77-4 possible. Triple-rising non-disposable Cyanogen Chloride material generates a lot of waste, which can 2,4-Dinitrophenol 51–28–5 be difficult to dispose of safely. -

2018 Treated Water Undetected Chemical Contaminant List
2018 Treated Water Undetected Chemical Contaminant List ESTROGENS AND OTHER HORMONES Diethylstilbestrol (DES) Estrone 17alpha-Estradiol 17alpha-Ethynal estradiol 17beta-Estradiol Progesterone Estriol cis-Testosterone trans-Testosterone INORGANIC CHEMICALS Antimony Niobium Arsenic Osmium Beryllium Palladium Cadmium Platinum Cerium Praseodymium Cesium Rhenium Cobalt Rhodium Cyanide Ruthenium Dysprosium Samarium Erbium Selenium Europium Silver Gadolinium Tantalum Gallium Tellurium Germanium Thallium Gold Thorium Hafnium Thulium Holmium Tin Iridium Titanium Lanthanum Tungsten Lead Uranium Lutetium Vanadium Mercury Ytterbium Molybdenum Zinc Neodymium Zirconium Nickel NITROSAMINES N-Nitropyrrolidine (NPYR) N-Nitrosomorpholine (NMOR) N-Nitrosodi-N-butylamine (NDBA) N-Nitrosodiphenylamine (NDPhA) N-Nitrosodiethylamine (NDEA) N-Nitrosodi-N-propylamine (NDPA) N-Nitrosodimethylamine (NDMA) N-Nitrosomethylethylamine (NMEA) N-Nitrosopiperidine (NPIP) 1 ORGANIC CHEMICALS Acenaphthene Butylbenzylphthalate Acenaphthylene Butyraldehyde (Butanal) Acetaldehyde Carbaryl Acetochlor Carbofuran Acetone Carbon disulfide Acrylamide Carbophenothion Acrylonitrile Carbon tetrachloride Alachlor Carboxin Aldicarb (Temik) Chlordane Aldicarb sulfone Chlordane, alpha Aldicarb sulfoxide Chlordane, gamma Aldrin Chlorfenvinphos Allyl chloride Chloroacetonitrile Tert-Amyl Methyl ether Chlorobenzene Ametryn Chlorobenzilate Anilizine 2-Chlorobiphenyl Anthracene 1-Chlorobutane Aspon Chloroethane Atraton Chloromethane Atrazine Chloroneb Azinphos-ethyl Chloroprene Azinphos-methyl -
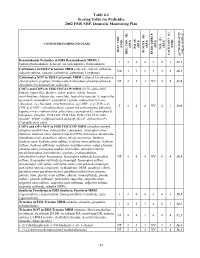
2002 NRP Section 6, Tables 6.1 Through
Table 6.1 Scoring Table for Pesticides 2002 FSIS NRP, Domestic Monitoring Plan } +1 0.05] COMPOUND/COMPOUND CLASS * ) (EPA) (EPA) (EPA) (EPA) (EPA) (FSIS) (FSIS) PSI (P) TOX.(T) L-1 HIST. VIOL. BIOCON. (B) {[( (2*R+P+B)/4]*T} REG. CON. (R) * ENDO. DISRUP. LACK INFO. (L) LACK INFO. {[ Benzimidazole Pesticides in FSIS Benzimidazole MRM (5- 131434312.1 hydroxythiabendazole, benomyl (as carbendazim), thiabendazole) Carbamates in FSIS Carbamate MRM (aldicarb, aldicarb sulfoxide, NA44234416.1 aldicarb sulfone, carbaryl, carbofuran, carbofuran 3-hydroxy) Carbamates NOT in FSIS Carbamate MRM (carbaryl 5,6-dihydroxy, chlorpropham, propham, thiobencarb, 4-chlorobenzylmethylsulfone,4- NT 4 1 3 NV 4 4 13.8 chlorobenzylmethylsulfone sulfoxide) CHC's and COP's in FSIS CHC/COP MRM (HCB, alpha-BHC, lindane, heptachlor, dieldrin, aldrin, endrin, ronnel, linuron, oxychlordane, chlorpyrifos, nonachlor, heptachlor epoxide A, heptachlor epoxide B, endosulfan I, endosulfan I sulfate, endosulfan II, trans- chlordane, cis-chlordane, chlorfenvinphos, p,p'-DDE, p, p'-TDE, o,p'- 3444NV4116.0 DDT, p,p'-DDT, carbophenothion, captan, tetrachlorvinphos [stirofos], kepone, mirex, methoxychlor, phosalone, coumaphos-O, coumaphos-S, toxaphene, famphur, PCB 1242, PCB 1248, PCB 1254, PCB 1260, dicofol*, PBBs*, polybrominated diphenyl ethers*, deltamethrin*) (*identification only) COP's and OP's NOT in FSIS CHC/COP MRM (azinphos-methyl, azinphos-methyl oxon, chlorpyrifos, coumaphos, coumaphos oxon, diazinon, diazinon oxon, diazinon met G-27550, dichlorvos, dimethoate, dimethoate -

Chemical Name Federal P Code CAS Registry Number Acutely
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extremely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extremely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extremely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extremely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extremely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extremely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extremely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extremely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extremely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime.