Identification of a Novel Selective Agonist of Pparc with No Promotion
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rosiglitazone-Associated Fractures in Type 2 Diabetes an Analysis from a Diabetes Outcome Progression Trial (ADOPT)
Clinical Care/Education/Nutrition/Psychosocial Research ORIGINAL ARTICLE Rosiglitazone-Associated Fractures in Type 2 Diabetes An analysis from A Diabetes Outcome Progression Trial (ADOPT) 1 7 STEVEN E. KAHN, MB, CHB DAHONG YU, PHD preclinical data and better understand the 2 7 BERNARD ZINMAN, MD MARK A. HEISE, PHD clinical implications of and possible interven- 3 7 JOHN M. LACHIN, SCD R. PAUL AFTRING, MD, PHD tions for these findings. 4 8 STEVEN M. HAFFNER, MD GIANCARLO VIBERTI, MD 5 WILLIAM H. HERMAN, MD FOR THE ADIABETES OUTCOME Diabetes Care 31:845–851, 2008 6 RURY R. HOLMAN, MD PROGRESSION TRIAL (ADOPT) STUDY 7 BARBARA G. KRAVITZ, MS GROUP* ype 2 diabetes is associated with an increased risk of fractures, with the risk increasing with longer duration OBJECTIVE — The purpose of this study was to examine possible factors associated with the T increased risk of fractures observed with rosiglitazone in A Diabetes Outcome Progression Trial of disease (1,2). These fractures affect pre- (ADOPT). dominantly the hip, arm, and foot (1–5) and occur despite the fact that bone min- RESEARCH DESIGN AND METHODS — Data from the 1,840 women and 2,511 men eral density is either normal or even in- randomly assigned in ADOPT to rosiglitazone, metformin, or glyburide for a median of 4.0 years creased in patients with type 2 diabetes were examined with respect to time to first fracture, rates of occurrence, and sites of fractures. compared with those who are not hyper- glycemic (5–7). Although the reason for RESULTS — In men, fracture rates did not differ between treatment groups. -

Diabetes Mellitus: Patterns of Pharmaceutical Use in Manitoba
Diabetes Mellitus: Patterns of Pharmaceutical Use in Manitoba by Kim¡ T. G. Guilbert A Thesis submitted to The Faculty of Graduate Studies in Partial Fulfillment of the Requirements for the Degree of MASTER OF SCIENCE Faculty of Pharmacy The University of Manitoba Winnipeg, Manitoba @ Kimi T.G. Guilbert, March 2005 TIIE UMYERSITY OF MANITOBA F'ACULTY OF GRADUATE STTJDIES +g+ù+ COPYRIGIIT PERMISSION PAGE Diabetes Mellitus: Patterns of Pharmaceutical Use in Manitoba BY Kimi T.G. Guilbert A ThesisÆracticum submitted to the Faculty of Graduate Studies of The University of Manitoba in partial fulfillment of the requirements of the degree of MASTER OF SCIENCE KIMI T.G. GTIILBERT O2()O5 Permission has been granted to the Library of The University of Manitoba to lend or sell copies of this thesis/practicum, to the National Library of Canada to microfïlm this thesis and to lend or sell copies of the film, and to University Microfilm Inc. to publish an abstract of this thesis/practicum. The author reserves other publication rights, and neither this thesis/practicum nor extensive extracts from it may be printed or otherwise reproduced without the author's written permission. Acknowledgements Upon initiation of this project I had a clear objective in mind--to learn more. As with many endeavors in life that are worthwhile, the path I have followed has brought me many places I did not anticipate at the beginning of my journey. ln reaching the end, it is without a doubt that I did learn more, and the knowledge I have been able to take with me includes a wider spectrum than the topic of population health and medication utilization. -

AVANDIA (Rosiglitazone Maleate Tablets), for Oral Use Ischemic Cardiovascular (CV) Events Relative to Placebo, Not Confirmed in Initial U.S
HIGHLIGHTS OF PRESCRIBING INFORMATION ----------------------- WARNINGS AND PRECAUTIONS ----------------------- These highlights do not include all the information needed to use • Fluid retention, which may exacerbate or lead to heart failure, may occur. AVANDIA safely and effectively. See full prescribing information for Combination use with insulin and use in congestive heart failure NYHA AVANDIA. Class I and II may increase risk of other cardiovascular effects. (5.1) • Meta-analysis of 52 mostly short-term trials suggested a potential risk of AVANDIA (rosiglitazone maleate tablets), for oral use ischemic cardiovascular (CV) events relative to placebo, not confirmed in Initial U.S. Approval: 1999 a long-term CV outcome trial versus metformin or sulfonylurea. (5.2) • Dose-related edema (5.3) and weight gain (5.4) may occur. WARNING: CONGESTIVE HEART FAILURE • Measure liver enzymes prior to initiation and periodically thereafter. Do See full prescribing information for complete boxed warning. not initiate therapy in patients with increased baseline liver enzyme levels ● Thiazolidinediones, including rosiglitazone, cause or exacerbate (ALT >2.5X upper limit of normal). Discontinue therapy if ALT levels congestive heart failure in some patients (5.1). After initiation of remain >3X the upper limit of normal or if jaundice is observed. (5.5) AVANDIA, and after dose increases, observe patients carefully for signs • Macular edema has been reported. (5.6) and symptoms of heart failure (including excessive, rapid weight gain; • Increased incidence of bone fracture was observed in long-term trials. dyspnea; and/or edema). If these signs and symptoms develop, the heart (5.7) failure should be managed according to current standards of care. -

AVANDIA® (Rosiglitazone Maleate) Tablets
PRESCRIBING INFORMATION AVANDIA® (rosiglitazone maleate) Tablets WARNING: CONGESTIVE HEART FAILURE ● Thiazolidinediones, including rosiglitazone, cause or exacerbate congestive heart failure in some patients (see WARNINGS). After initiation of AVANDIA, and after dose increases, observe patients carefully for signs and symptoms of heart failure (including excessive, rapid weight gain, dyspnea, and/or edema). If these signs and symptoms develop, the heart failure should be managed according to current standards of care. Furthermore, discontinuation or dose reduction of AVANDIA must be considered. ● AVANDIA is not recommended in patients with symptomatic heart failure. Initiation of AVANDIA in patients with established NYHA Class III or IV heart failure is contraindicated. (See CONTRAINDICATIONS and WARNINGS.) DESCRIPTION AVANDIA (rosiglitazone maleate) is an oral antidiabetic agent which acts primarily by increasing insulin sensitivity. AVANDIA is used in the management of type 2 diabetes mellitus (also known as non-insulin-dependent diabetes mellitus [NIDDM] or adult-onset diabetes). AVANDIA improves glycemic control while reducing circulating insulin levels. Pharmacological studies in animal models indicate that rosiglitazone improves sensitivity to insulin in muscle and adipose tissue and inhibits hepatic gluconeogenesis. Rosiglitazone maleate is not chemically or functionally related to the sulfonylureas, the biguanides, or the alpha-glucosidase inhibitors. Chemically, rosiglitazone maleate is (±)-5-[[4-[2-(methyl-2- pyridinylamino)ethoxy]phenyl]methyl]-2,4-thiazolidinedione, (Z)-2-butenedioate (1:1) with a molecular weight of 473.52 (357.44 free base). The molecule has a single chiral center and is present as a racemate. Due to rapid interconversion, the enantiomers are functionally indistinguishable. The structural formula of rosiglitazone maleate is: The molecular formula is C18H19N3O3S•C4H4O4. -

Utah Medicaid Pharmacy and Therapeutics Committee Drug
Utah Medicaid Pharmacy and Therapeutics Committee Drug Class Review DPP-4 Inhibitor Products AHFS Classification: 68:20.05 Dipeptidyl Peptidase-4 Inhibitors Alogliptin (Nesina) Alogliptin and Metformin (Kazano) Alogliptin and Pioglitazone (Oseni) Linagliptin (Tradjenta) Linagliptin and Empagliflozin (Glyxambi) Linagliptin and Metformin (Jentadueto, Jentadueto XR) Saxagliptin (Onglyza) Saxagliptin and Dapagliflozin (Qtern) Saxagliptin and Metformin (Kombiglyze XR) Sitagliptin (Januvia) Sitagliptin and Metformin (Janumet, Janumet XR) Final Report November 2017 Review prepared by: Elena Martinez Alonso, B.Pharm., MSc MTSI, Medical Writer Valerie Gonzales, Pharm.D., Clinical Pharmacist Vicki Frydrych, Pharm.D., Clinical Pharmacist Joanita Lake, B.Pharm., MSc EBHC (Oxon), Assistant Professor University of Utah College of Pharmacy Michelle Fiander, MA, MLIS, Systematic Review/Evidence Synthesis Librarian Joanne LaFleur, Pharm.D., MSPH, Associate Professor University of Utah College of Pharmacy University of Utah College of Pharmacy, Drug Regimen Review Center Copyright © 2017 by University of Utah College of Pharmacy Salt Lake City, Utah. All rights reserved 1 Contents List of Abbreviations .................................................................................................................................... 3 Executive Summary ...................................................................................................................................... 4 Introduction .................................................................................................................................................. -

Muraglitazar Bristol-Myers Squibb/Merck Daniella Barlocco
Muraglitazar Bristol-Myers Squibb/Merck Daniella Barlocco Address Originator Bristol-Myers Squibb Co University of Milan . Istituto di Chimica Farmaceutica e Tossicologica Viale Abruzzi 42 Licensee Merck & Co Inc 20131 Milano . Italy Status Pre-registration Email: [email protected] . Indications Metabolic disorder, Non-insulin-dependent Current Opinion in Investigational Drugs 2005 6(4): diabetes © The Thomson Corporation ISSN 1472-4472 . Actions Antihyperlipidemic agent, Hypoglycemic agent, Bristol-Myers Squibb and Merck & Co are co-developing Insulin sensitizer, PPARα agonist, PPARγ agonist muraglitazar, a dual peroxisome proliferator-activated receptor-α/γ . agonist, for the potential treatment of type 2 diabetes and other Synonym BMS-298585 metabolic disorders. In November 2004, approval was anticipated as early as mid-2005. Registry No: 331741-94-7 Introduction [579218], [579221], [579457], [579459]. PPARγ is expressed in Type 2 diabetes is a complex metabolic disorder that is adipose tissue, lower intestine and cells involved in characterized by hyperglycemia, insulin resistance and immunity. Activation of PPARγ regulates glucose and lipid defects in insulin secretion. The disease is associated with homeostasis, and triggers insulin sensitization [579216], older age, obesity, a family history of diabetes and physical [579218], [579458], [579461]. PPARδ is expressed inactivity. The prevalence of type 2 diabetes is increasing ubiquitously and has been found to be effective in rapidly, and the World Health Organization warns that, controlling dyslipidemia and cardiovascular diseases unless appropriate action is taken, the number of sufferers [579216]. PPARα agonists are used as potent hypolipidemic will double to over 350 million individuals by the year compounds, increasing plasma high-density lipoprotein 2030. Worryingly, it is estimated that only half of sufferers (HDL)-cholesterol and reducing free fatty acids, are diagnosed with the condition [www.who.int]. -

Thiazolidinedione Derivatives in Type 2 Diabetes Mellitus
r E v i E w Thiazolidinedione derivatives in type 2 diabetes mellitus C.J.J. Tack, P. Smits*# Department of Internal Medicine, Radboud University Nijmegen Medical Centre, Nijmegen, the Netherlands, *corresponding author: [email protected] A b s T r act in Europe, the thiazolidinedione derivatives pioglitazone approved for use in combination therapy. Unlike the situation and rosiglitazone have been approved for the treatment of in the USA, TZDs are not approved, but even contraindicated type 2 diabetes mellitus either as monotherapy for patients for use in combination with insulin in Europe. From the day with intolerance or contraindications to metformin or in of approval onwards, there has been discussion concerning combination therapy. This class of drugs seems particularly the exact place of TZDs within the pharmacotherapy of suited for obese patients, but is currently not considered as type 2 diabetes mellitus. Different views have resulted in a first choice for monotherapy. The efficacy with respect to differences in guidelines and treatment standards. The lack blood glucose lowering is comparable with sulphonylurea of data on long-term clinical studies with ‘hard’ endpoints (SU) derivatives and with metformin. long-term data with (mortality and new macrovascular events) definitively plays respect to efficacy and side effects are still limited. an important role in this discussion. Recently, the first outcome study was published (the PROactive study),1 but this study has also raised several questions.2-5 K E y w o r d s With respect to glucose regulation, TZDs do not seem to be superior to the conventional drugs metformin or Combination therapy, monotherapy, pioglitazone, sulphonylurea (SU) derivatives. -

Thiazolidinedione Drugs Down-Regulate CXCR4 Expression on Human Colorectal Cancer Cells in a Peroxisome Proliferator Activated Receptor Á-Dependent Manner
1215-1222 26/3/07 18:16 Page 1215 INTERNATIONAL JOURNAL OF ONCOLOGY 30: 1215-1222, 2007 Thiazolidinedione drugs down-regulate CXCR4 expression on human colorectal cancer cells in a peroxisome proliferator activated receptor Á-dependent manner CYNTHIA LEE RICHARD and JONATHAN BLAY Department of Pharmacology, Faculty of Medicine, Dalhousie University, Halifax, Nova Scotia, B3H 1X5, Canada Received October 26, 2006; Accepted December 4, 2006 Abstract. Peroxisome proliferator activated receptor (PPAR) Introduction Á is a nuclear receptor involved primarily in lipid and glucose metabolism. PPARÁ is also expressed in several cancer types, Peroxisome proliferator activated receptors (PPARs) are and has been suggested to play a role in tumor progression. nuclear hormone receptors that are involved primarily in PPARÁ agonists have been shown to reduce the growth of lipid and glucose metabolism (1). Upon ligand activation, colorectal carcinoma cells in culture and in xenograft models. these receptors interact with the retinoid X receptor (RXR) Furthermore, the PPARÁ agonist thiazolidinedione has been and bind to peroxisome proliferator response elements (PPREs), shown to reduce metastasis in a murine model of rectal cancer. leading to transcriptional regulation of target genes. Members Since the chemokine receptor CXCR4 has emerged as an of the thiazolidinedione class of antidiabetic drugs act as important player in tumorigenesis, particularly in the process ligands for PPARÁ (2), as does endogenously produced 15- 12,14 of metastasis, we sought to determine if PPARÁ agonists deoxy-¢ -prostaglandin J2 (15dPGJ2) (3). might act in part by reducing CXCR4 expression. We found In addition to regulation of glucose metabolism, PPARÁ that rosiglitazone, a thiazolidinedione PPARÁ agonist used appears also to be involved in tumorigenesis, although its primarily in the treatment of type 2 diabetes, significantly exact role has yet to be elucidated (4). -
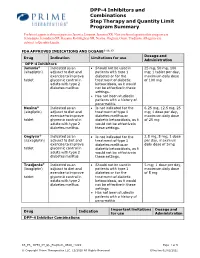
DPP-4 Inhibitors and Combinations Step Therapy and Quantity Limit
DPP-4 Inhibitors and Combinations Step Therapy and Quantity Limit Program Summary Preferred agents in this program are Januvia, Janumet, Janumet XR. Non-preferred agents in this program are Jentadueto, Jentadueto XR, Kazano, Kombiglyze XR, Nesina, Onglyza, Oseni, Tradjenta. All agents are subject to Quantity Limits. FDA APPROVED INDICATIONS AND DOSAGE2-11, 13 Dosage and Drug Indication Limitations for use Administration DPP-4 Inhibitors Januvia® Indicated as an • Should not be used in 25 mg, 50 mg, 100 (sitagliptin) adjunct to diet and patients with type 1 mg; 1 tablet per day, exercise to improve diabetes or for the maximum daily dose tablet glycemic control in treatment of diabetic of 100 mg adults with type 2 ketoacidosis, as it would diabetes mellitus not be effective in these settings. • Has not been studied in patients with a history of pancreatitis. Nesina® Indicated as an • Is not indicated for the 6.25 mg, 12.5 mg, 25 (alogliptin) adjunct to diet and treatment of type 1 mg; 1 dose per day, exercise to improve diabetes mellitus or maximum daily dose tablet glycemic control in diabetic ketoacidosis, as it of 25 mg adults with type 2 would not be effective in diabetes mellitus these settings. Onglyza® Indicated as an • Is not indicated for the 2.5 mg, 5 mg; 1 dose (saxagliptin) adjunct to diet and treatment of type 1 per day, maximum exercise to improve diabetes mellitus or daily dose of 5 mg tablet glycemic control in diabetic ketoacidosis, as it adults with type 2 would not be effective in diabetes mellitus these settings. -

Pioglitazone, a Peroxisome Proliferator-Activated Receptor
0023-6837/03/8312-1715$03.00/0 LABORATORY INVESTIGATION Vol. 83, No. 12, p. 1715, 2003 Copyright © 2003 by The United States and Canadian Academy of Pathology, Inc. Printed in U.S.A. Pioglitazone, a Peroxisome Proliferator-Activated Receptor-␥ Agonist, Attenuates Myocardial Ischemia/Reperfusion Injury in a Rat Model Haruyasu Ito, Atsushi Nakano, Makoto Kinoshita, and Akira Matsumori The Department of Cardiovascular Medicine, Kyoto University, Graduate School of Medicine, Kyoto, Japan SUMMARY: Thiazolidinediones are insulin-sensitizing drugs, ligands for peroxisome proliferator-activated receptor-␥ (PPAR-␥), which play an important role in the modulation of inflammatory responses. Myocardial ischemia/reperfusion (MI/R) injury is associated with inflammation, in which various cells, particularly monocytes and macrophages, are involved. This study examined the effects of the thiazolidinedione peroxisome proliferator-activated receptor-␥ ligand, pioglitazone, in a rat model of MI/R injury. Pioglitazone at 3 mg/kg/day or the vehicle was administered for 7 days before rats were subjected to 30 minutes of coronary ligation followed by 24 hours of reperfusion. The mRNA expression [monocyte chemoattractant protein-1 (MCP-1) and intercellular adhesion molecule-1] in the ischemic region, the number of infiltrating macrophages in the ischemic region, and the myocardial infarct size were examined. The inhibitory effects of pioglitazone on activated macrophages were studied in vitro. Phorbol 12-myristate 13-acetate–induced MCP-1 production, in the absence or presence of pioglitazone, were assayed in cultured macrophages. Compared with the control group, (1) mRNA levels of MCP-1 and intercellular adhesion molecule-1 and the number of infiltrating macrophages in the ischemic region were significantly lower in the pioglitazone-treated group; and (2) myocardial infarct size was significantly smaller in the pioglitazone-treated group. -

Peroxisome Proliferator-Activated Receptors and Their Novel Ligands As Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease
cells Review Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease Anne Fougerat 1,*, Alexandra Montagner 1,2,3, Nicolas Loiseau 1 , Hervé Guillou 1 and Walter Wahli 1,4,5,* 1 Institut National de la Recherche Agronomique (INRAE), ToxAlim, UMR1331 Toulouse, France; [email protected] (A.M.); [email protected] (N.L.); [email protected] (H.G.) 2 Institut National de la Santé et de la Recherche Médicale (Inserm), Institute of Metabolic and Cardiovascular Diseases, UMR1048 Toulouse, France 3 Institute of Metabolic and Cardiovascular Diseases, University of Toulouse, UMR1048 Toulouse, France 4 Lee Kong Chian School of Medicine, Nanyang Technological University Singapore, Clinical Sciences Building, 11 Mandalay Road, Singapore 308232, Singapore 5 Center for Integrative Genomics, Université de Lausanne, Le Génopode, CH-1015 Lausanne, Switzerland * Correspondence: [email protected] (A.F.); [email protected] (W.W.); Tel.: +33-582066376 (A.F.); +41-79-6016832 (W.W.) Received: 29 May 2020; Accepted: 4 July 2020; Published: 8 July 2020 Abstract: Non-alcoholic fatty liver disease (NAFLD) is a major health issue worldwide, frequently associated with obesity and type 2 diabetes. Steatosis is the initial stage of the disease, which is characterized by lipid accumulation in hepatocytes, which can progress to non-alcoholic steatohepatitis (NASH) with inflammation and various levels of fibrosis that further increase the risk of developing cirrhosis and hepatocellular carcinoma. The pathogenesis of NAFLD is influenced by interactions between genetic and environmental factors and involves several biological processes in multiple organs. No effective therapy is currently available for the treatment of NAFLD. -

Pharmacy Medical Necessity Guidelines: Thiazolidinediones
Pharmacy Medical Necessity Guidelines: Thiazolidinediones (TZDs) Effective: March 9, 2021 Prior Authorization Required √ Type of Review – Care Management Not Covered Type of Review – Clinical Review √ Pharmacy (RX) or Medical (MED) Benefit RX Department to Review RXUM These pharmacy medical necessity guidelines apply to the following: Fax Numbers: Commercial Products RXUM: 617.673.0988 Tufts Health Plan Commercial products – large group plans Tufts Health Plan Commercial products – small group and individual plans Tufts Health Freedom Plan products – large group plans Tufts Health Freedom Plan products – small group plans • CareLinkSM – Refer to CareLink Procedures, Services and Items Requiring Prior Authorization Tufts Health Public Plans Products Tufts Health Direct – A Massachusetts Qualified Health Plan (QHP) (a commercial product) Tufts Health Together – MassHealth MCO Plan and Accountable Care Partnership Plans Tufts Health RITogether – A Rhode Island Medicaid Plan Note: This guideline does not apply to Medicare Members (includes dual eligible Members). OVERVIEW FOOD AND DRUG ADMINISTRATION-APPROVED INDICATIONS ActoPlus Met XR (pioglitazone/metformin extended release tablet) is a thiazolidinedione and biguanide combination product indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus when treatment with both pioglitazone and metformin is appropriate. Avandia (rosiglitazone) is a thiazolidinedione indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. Neither Avandia nor ActoPlus Met XR is indicated for the treatment of type 1 diabetes or diabetic ketoacidosis. Thiazolidinediones (TZDs) have a Black Box Warning regarding heart failure; TZDs have been associated with causing or exacerbating congestive heart failure.