Prof. Samia El-Azab 2017
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

59. Lateral Facial Clefts
59 LATERAL FACIAL CLEFTS LI OR TRANSVERSE CLEFTS ARE CONSIDERED THE RESULT OF FAILURE OF MESODERM MIGRATION OR MERGING TO OBLITERATE MANDIBULAR THE EMBRYONIC GROOVES BETWEEN THE MAXILLARY AND PROMINENCES TRANSVERSE CLEFTS AS THESE CLEFTS ARE RARE AND ALMOST EVERYBODY HAVING ONE HAS AND REPORTED IT IT IS POSSIBLE TO REVIEW MOST OF THE REPORTED CASES 769 DESCRIBED THE AFTER WHEN NOTE TREATMENT SPECIFIC CASE RECORDINGS IN WHAT MAY SEEM HELTERSKELTER ARRANGEMENT GENERALIZATIONS MAY BE OF VALUE IN 1891 ROSE NOTED FOR LONG THE VERY EXISTENCE OF THIS MACROSROMATOUS DEFORMITY WAS DOUBTED BUT CASES HAVE BEEN RECOGNIZED MORE OR LESS SINCE 1715 WHEN MURALT PICTURED IT FOR THE FIRST TIME ONE OF THE FIRST CASES REPORTED WAS BY VROLIK WHOIN HIS 1849 CLEFTS WORK GAVE SEVERAL ILLUSTRATIONS OF COMMISSURAL AS WELL AS OTHER DEFORMITIES OF THE FACE OTHER CASES WERE REPORTED BY REISSMANN IN 1869 AND MORGAN IN 1882 MACROSTOMIA OR COMMISSURAL HARELIP ACCORDING TO ROSE IS DIAMETER OF WHICH EVIDENCED BY AN INCREASED THE MOUTH MAY VARY IN FROM SLIGHT INCREASE TO CONSIDERABLE DISTANCE CASE RE PORTED BY RYND IN 1862 THE MOUTH OPENING EXTENDED AS FAR AS THE THE LEFT FIRST MOLAR ON THE RIGHT SIDE AND TO THE LAST MOLAR ON IN 1887 SUTTON PUBLISHED THE DRAWING OF CHILD WITH VERY LARGE RED CICATRIX THIS CLEFT THE ANGLES OF WHICH GRADUALLY PASSED INTO SCAR ENDED IN GAPING WOUND OVER THE TEMPORAL REGION EXTEND ING TO THE DURA MATER ROSE ALSO POINTED OUT MACROSROMA IS NOR ONLY ATTENDED BY GREAT DISFIGUREMENT HUT IS ALSO TROU BLESOME FROM THE IMPOSSIBILITY OF THE CHILD RETAINING -

OCSHCN-10G, Medical Eligibility List for Clinical and Case Management Services.Pdf
OCSHCN-10g (01 2019) (Rev 7-15-2017) Office for Children with Special Health Care Needs Medical Eligibility List for Clinical and Case Management Services BODY SYSTEM ELIGIBLE DISEASES/CONDITIONS ICD-10-CM CODES AFFECTED AUTISM SPECTRUM Autistic disorder, current or active state F84.0 Autistic disorder DISORDER (ASD) F84.3 Other childhood disintegrative disorder Autistic disorder, residual state F84.5 Asperger’s Syndrome F84.8 Other pervasive developmental disorder Other specified pervasive developmental disorders, current or active state Other specified pervasive developmental disorders, residual state Unspecified pervasive development disorder, current or active Unspecified pervasive development disorder, residual state CARDIOVASCULAR Cardiac Dysrhythmias I47.0 Ventricular/Arrhythmia SYSTEM I47.1 Supraventricular/Tachycardia I47.2 Ventricular/Tachycardia I47.9 Paroxysmal/Tachycardia I48.0 Paroxysmal atrial fibrillation I48.1 Persistent atrial fibrillationar I48.2 Chronic atrial fibrillation I48.3 Typical atrial flutter I48.4 Atypical atrial flutter I49.0 Ventricular fibrillation and flutter I49.1 Atrial premature depolarization I49.2 Junctional premature depolarization I49.3 Ventricular premature depolarization I49.49 Ectopic beats Extrasystoles Extrasystolic arrhythmias Premature contractions Page 1 of 28 OCSHCN-10g (01 2019) (Rev 7-15-2017) Office for Children with Special Health Care Needs Medical Eligibility List for Clinical and Case Management Services I49.5 Tachycardia-Bradycardia Syndrome CARDIOVASCULAR Chronic pericarditis -

MICHIGAN BIRTH DEFECTS REGISTRY Cytogenetics Laboratory Reporting Instructions 2002
MICHIGAN BIRTH DEFECTS REGISTRY Cytogenetics Laboratory Reporting Instructions 2002 Michigan Department of Community Health Community Public Health Agency and Center for Health Statistics 3423 N. Martin Luther King Jr. Blvd. P. O. Box 30691 Lansing, Michigan 48909 Michigan Department of Community Health James K. Haveman, Jr., Director B-274a (March, 2002) Authority: P.A. 236 of 1988 BIRTH DEFECTS REGISTRY MICHIGAN DEPARTMENT OF COMMUNITY HEALTH BIRTH DEFECTS REGISTRY STAFF The Michigan Birth Defects Registry staff prepared this manual to provide the information needed to submit reports. The manual contains copies of the legislation mandating the Registry, the Rules for reporting birth defects, information about reportable and non reportable birth defects, and methods of reporting. Changes in the manual will be sent to each hospital contact to assist in complete and accurate reporting. We are interested in your comments about the manual and any suggestions about information you would like to receive. The Michigan Birth Defects Registry is located in the Office of the State Registrar and Division of Health Statistics. Registry staff can be reached at the following address: Michigan Birth Defects Registry 3423 N. Martin Luther King Jr. Blvd. P.O. Box 30691 Lansing MI 48909 Telephone number (517) 335-8678 FAX (517) 335-9513 FOR ASSISTANCE WITH SPECIFIC QUESTIONS PLEASE CONTACT Glenn E. Copeland (517) 335-8677 Cytogenetics Laboratory Reporting Instructions I. INTRODUCTION This manual provides detailed instructions on the proper reporting of diagnosed birth defects by cytogenetics laboratories. A report is required from cytogenetics laboratories whenever a reportable condition is diagnosed for patients under the age of two years. -

Prevalence and Incidence of Rare Diseases: Bibliographic Data
Number 1 | January 2019 Prevalence and incidence of rare diseases: Bibliographic data Prevalence, incidence or number of published cases listed by diseases (in alphabetical order) www.orpha.net www.orphadata.org If a range of national data is available, the average is Methodology calculated to estimate the worldwide or European prevalence or incidence. When a range of data sources is available, the most Orphanet carries out a systematic survey of literature in recent data source that meets a certain number of quality order to estimate the prevalence and incidence of rare criteria is favoured (registries, meta-analyses, diseases. This study aims to collect new data regarding population-based studies, large cohorts studies). point prevalence, birth prevalence and incidence, and to update already published data according to new For congenital diseases, the prevalence is estimated, so scientific studies or other available data. that: Prevalence = birth prevalence x (patient life This data is presented in the following reports published expectancy/general population life expectancy). biannually: When only incidence data is documented, the prevalence is estimated when possible, so that : • Prevalence, incidence or number of published cases listed by diseases (in alphabetical order); Prevalence = incidence x disease mean duration. • Diseases listed by decreasing prevalence, incidence When neither prevalence nor incidence data is available, or number of published cases; which is the case for very rare diseases, the number of cases or families documented in the medical literature is Data collection provided. A number of different sources are used : Limitations of the study • Registries (RARECARE, EUROCAT, etc) ; The prevalence and incidence data presented in this report are only estimations and cannot be considered to • National/international health institutes and agencies be absolutely correct. -

Goldenhar Syndrome
ndrom Sy es tic & e G n e e n G e f Saxena and David, J Genet Syndr Gene Ther 2012, 3:2 T o Journal of Genetic Syndromes h l e a r n a DOI: 10.4172/2157-7412.1000113 r p u y o J & Gene Therapy ISSN: 2157-7412 Case Report Open Access Goldenhar Syndrome - A Rare Case Report Runjhun Saxena1* and Maria Priscilla David2 1Department of Oral Medicine and Radiology, Karnavati School of Dentistry, Uvarsad, Gandhinagar, Gujarat 382422, India 2Department of Oral Medicine and Radiology, M R Ambedkar Dental College,1/36 Cline Road, Cooke Town, Bangalore-05, India Abstract Goldenhar syndrome or oculo-auriculovertebral (OAV) is a rare abnormality affecting the craniofacial region having extracranial manifestations as well. First described by Maurice Goldenhar, its etiology still remains uncertain. We describes a case of Goldenhar syndrome with craniofacial manifestations which makes it amenable to diagnosis by an oral physician. Keywords: Goldenhar syndrome; Mandibular hypoplasia; • Hands / Fingers: clubbing, polydactyly, clinodactyly, single Periauricular tags; Corneal opacities palmar crease Introduction • Vertebral column anomalies (atlas occipitalization, synosto- sis, hemivertebrae, fused vertebrae, scoliosis, and bifid spine) Franceschetti-Goldenhar syndrome or Goldenhar syndrome, also [4]. known as facioauriculovertebral spectrum (FAV), first and second branchial arch syndrome, or oculo-auriculovertebral (OAV) spectrum Principal deformities of the Goldenhar syndrome are often is a rare congenital malformation which encompasses various combined with various malformations, such as: morphological and functional abnormalities. The syndrome was first • Cleft lip and/or palate, tongue cleft, unilateral tongue recorded by German physician Carl Ferdinand Von Arlt in 1845, hypoplasia, and parotid gland aplasia. -

Back Matter 611-642.Pdf
Index A Acrodysostosis, 527, 538 Aarskog syndrome, 527, 537 Acrofacial dysostosis 1, 580–581 differentiated from Opitz G/BBB Acrofacial dysplasia, 563 syndrome, 458 Acro-fronto-facio-nasal dysostosis Aase-Smith syndrome, 527 syndrome, 527, 538–539 Aase syndrome, 527, 537 Acromegaloid facial appearance Abducens nerve. See Cranial nerve VI syndrome, 527, 539 Abduction, Möbius sequence-related Acromegaloid phenotype-cutis verticis impairment of, 197, 198 gyrata-cornea leukoma, 527, 539 Abetalipoproteinemia, 527, 537 Acromesomelic dysplasia, Maroteaux- Ablepharon-macrostomia syndrome, Martinelli-Campailla type, 527, 527, 538 539 Abortion, spontaneous, chromosomal Acromicric dysplasia, 527, 539 abnormalities associated with, 83 Acro-osteolysis syndrome, 565 Abruzzo-Erickson syndrome, 527, 538 Acro-reno-ocular syndrome, 527, Acanthosis, 607 539 Acanthosis nigricans, Crouzon Acyclovir, 504, 505, 518 syndrome-related, 169 Adam complex, 207–209 Physician-parent interactions, Adams-Oliver syndrome, 527, 540 69–70 Adduction, Möbius sequence-related Acetyl-coenzyme A deficiency, 364 impairment of, 197, 198 N-Acetylcystine, 321 Adenoma sebaceum, tuberous sclerosis N-Acetyl galactosamine-4-sulfatase complex-related, 304, 308–309 deficiency, 366 Adrenocorticotropic hormone, 433 N-Acetyl galactosamine-6-sulfatase Aging, premature. See also Progeria deficiency, 357, 365 ataxia-telangiectasia-related, 320 a-N-Acetyl glucosaminidase acetyl Aglossia-adactyly syndrome, 584–585 transferase deficiency, 357 Agnathia-holoprosencephaly, 54 a-N-Acetyl glucosaminidase -

Description Concept ID Synonyms Definition
Description Concept ID Synonyms Definition Category ABNORMALITIES OF TEETH 426390 Subcategory Cementum Defect 399115 Cementum aplasia 346218 Absence or paucity of cellular cementum (seen in hypophosphatasia) Cementum hypoplasia 180000 Hypocementosis Disturbance in structure of cementum, often seen in Juvenile periodontitis Florid cemento-osseous dysplasia 958771 Familial multiple cementoma; Florid osseous dysplasia Diffuse, multifocal cementosseous dysplasia Hypercementosis (Cementation 901056 Cementation hyperplasia; Cementosis; Cementum An idiopathic, non-neoplastic condition characterized by the excessive hyperplasia) hyperplasia buildup of normal cementum (calcified tissue) on the roots of one or more teeth Hypophosphatasia 976620 Hypophosphatasia mild; Phosphoethanol-aminuria Cementum defect; Autosomal recessive hereditary disease characterized by deficiency of alkaline phosphatase Odontohypophosphatasia 976622 Hypophosphatasia in which dental findings are the predominant manifestations of the disease Pulp sclerosis 179199 Dentin sclerosis Dentinal reaction to aging OR mild irritation Subcategory Dentin Defect 515523 Dentinogenesis imperfecta (Shell Teeth) 856459 Dentin, Hereditary Opalescent; Shell Teeth Dentin Defect; Autosomal dominant genetic disorder of tooth development Dentinogenesis Imperfecta - Shield I 977473 Dentin, Hereditary Opalescent; Shell Teeth Dentin Defect; Autosomal dominant genetic disorder of tooth development Dentinogenesis Imperfecta - Shield II 976722 Dentin, Hereditary Opalescent; Shell Teeth Dentin Defect; -

Developmental Anomalies of the Temporomandibular Joint
Developmental Anomalies of the Temporomandibular Joint R. Bruce Ross, DDS, MSc, FRCD he processes by which the human face develops in the Division of Orthodontics embryo are exceedingly complex, but they work out per- The Hospital for Sick Children fectly—-almost every time. Occasionally, however, the Toronto, Ontario, Canada T development of structures such as those comprising the temporo- mandibular articulation is disturbed, leading to an anomalous Professor Faoulty of Dentistry morphology in later life. It is important to note that an anomaly is University of Toronto not necessarily an undesirable condition requiring treatment. It Toronto, Ontario, Canada may be benign, with no associated problems, and therefore of no consequence; in fact, it may never be identified. Ross and Correspondence to: Johnston' have published a review of the developmental processes Dr R. B. Ross and etiology of undesirable conditions affecting the temporo- Faculty oS Dentistry mandibular joint (TMJ). University of Toronto Craniofacial anomalies may be caused by genetic faults (eg, 124 Edward Street Treacher Collins syndrome) or caused by a teratogenic agent (eg, Toronto, Ontario thahdomide), but most often it appears the cause is multifactorial M5G lG6 0anads Fax:416-813-6375 (eg, cleft hp and palate). When the entire human genome is avail- E-mail: [email protected] able and studied in persons with congenital anomalies, there may well be indicators for, or at least a demonstrable predisposition to, developmental problems. Problems involving the TMJ can be acquired or congenital (developmental). Tbe vast majority seen in a dental office are acquired dysfunctions, traumatic injuries, or pathologic condi- tions. -

EUROCAT Syndrome Guide
JRC - Central Registry european surveillance of congenital anomalies EUROCAT Syndrome Guide Definition and Coding of Syndromes Version July 2017 Revised in 2016 by Ingeborg Barisic, approved by the Coding & Classification Committee in 2017: Ester Garne, Diana Wellesley, David Tucker, Jorieke Bergman and Ingeborg Barisic Revised 2008 by Ingeborg Barisic, Helen Dolk and Ester Garne and discussed and approved by the Coding & Classification Committee 2008: Elisa Calzolari, Diana Wellesley, David Tucker, Ingeborg Barisic, Ester Garne The list of syndromes contained in the previous EUROCAT “Guide to the Coding of Eponyms and Syndromes” (Josephine Weatherall, 1979) was revised by Ingeborg Barisic, Helen Dolk, Ester Garne, Claude Stoll and Diana Wellesley at a meeting in London in November 2003. Approved by the members EUROCAT Coding & Classification Committee 2004: Ingeborg Barisic, Elisa Calzolari, Ester Garne, Annukka Ritvanen, Claude Stoll, Diana Wellesley 1 TABLE OF CONTENTS Introduction and Definitions 6 Coding Notes and Explanation of Guide 10 List of conditions to be coded in the syndrome field 13 List of conditions which should not be coded as syndromes 14 Syndromes – monogenic or unknown etiology Aarskog syndrome 18 Acrocephalopolysyndactyly (all types) 19 Alagille syndrome 20 Alport syndrome 21 Angelman syndrome 22 Aniridia-Wilms tumor syndrome, WAGR 23 Apert syndrome 24 Bardet-Biedl syndrome 25 Beckwith-Wiedemann syndrome (EMG syndrome) 26 Blepharophimosis-ptosis syndrome 28 Branchiootorenal syndrome (Melnick-Fraser syndrome) 29 CHARGE -

Physical Examination of Newborn by Dr Behzad Barekatain MD Assistant Professor of Pediatrics, Neonatologist Academic Member of Isfahan University of Medical Sciences
Physical examination of Newborn By Dr behzad barekatain MD Assistant professor of pediatrics, neonatologist Academic member of isfahan university of medical sciences 1 Swelling of the eyelids is common in newborns and resolves during the first few days of life. Subconjunctival hemorrhages are common. They occur during delivery and resolve in 1 to 2 weeks. 2 The normally white sclera is evaluated for changes of color. A bluish coloration, however, is present in premature infants and in other small babies because of their very thin sclera.also presents in baby with osteogenesis imperfecta. There may also be a mucoid discharge affecting the eyes, often called a “sticky eye,” in the first few days of life, which resolves spontaneously; if more prolonged, it is often from a blocked or incompletely canalized nasolacrimal duct. The eyelids can be cleansed with sterile water. This condition must be contrasted with the erythematous, swollen eyelids with purulent eye discharge seen in conjunctivitis. 4 5 ABNORMAL RED REFLEX This is one of the most important abnormalities that requires immediate evaluation. The term leukocoria is used to describe a white pupil seen by the naked eye or during the red reflex test. Leukocoria is not a diagnosis but rather a description of an observation. Because the infant sleeps much of the time and because the pupils are small, a white pupil often is not noticed until the infant becomes more alert and active. False-positive red reflex test results are commonly due to small pupils, shifting gaze, limited patient cooperation, poor illumination from the ophthalmoscope, and examiner inexperience. -

Síndrome De Treacher Collins: Revisão De Literatura
Argito de Revisão Síndrome de Treacher Collins: Revisão de Literatura Treacher Collins Syndrome: Review of the Literature Dorivaldo Lopes da Silva*, Francisco Xavier Palheta Neto**, Stéphanie Gonçalves Carneiro***, Kelly Leticia Castro Souza****, Suellen da Silva Souza****, Angélica Cristina Pezzin Palheta*****. * Monitor da Disciplina de Otorrinolaringologia da Universidade do Estado do Pará. Aluno do sexto ano do Curso de Medicina. ** Professor Assistente da Universidade do Estado do Pará. Coordenador Científico e Preceptor da Residência Médica em Otorrinolaringologia do Hospital Universitário Bettina Ferro de Souza da Universidade Federal do Pará. *** Monitora da Disciplina de Oftalmologia da Universidade do Estado do Pará. Aluna do sexto ano do Curso de Medicina. **** Aluna do Quinto ano do Curso de Medicina. Universidade do Estado do Pará. ***** Professora Assistente da Universidade do Estado do Pará. Coordenadora do Programa de Saúde Auditiva do Hospital Universitário Bettina Ferro de Souza da Universidade Federal do Pará. Instituição: Universidade do Estado do Pará. Endereço para correspondênciA: Dorivaldo Lopes da Silva – Trav. Dom Romualdo de Seixas, 1630 – Apto 404 – Belém / PA – CEP: 66055-200 – Telefone: (91) 3225- 0158 / (91) 9185-2065 – E-mail: [email protected] Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da R@IO em 23 de dezembro de 2007. Cod. 397. Artigo aceito em 1 de fevereiro de 2008. RESUMO Introdução: A Síndrome de Treacher Collins é um distúrbio hereditário caracterizado por anomalias craniofaciais e manifesta- se com diversas variáveis clínicas apresentando incidência aproximada de 1:40.000 a 1:70.000 pessoas. Objetivo: Realizar uma revisão de literatura sobre os diversos aspectos da Síndrome de Treacher Collins, enfatizando as manifestações clínicas otorrinolaringológicas. -
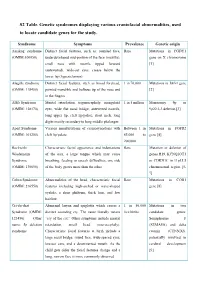
S2 Table. Genetic Syndromes Displaying Various Craniofacial Abnormalities, Used to Locate Candidate Genes for the Study
S2 Table. Genetic syndromes displaying various craniofacial abnormalities, used to locate candidate genes for the study. Syndrome Symptoms Prevalence Genetic origin Aarskog syndrome Distinct facial features, such as: rounded face, Rare Mutations in FGDY1 (OMIM:100050) underdeveloped mid-portion of the face (maxilla), gene on X chromosome small nose with nostrils tipped forward [1] (anteverted), wide-set eyes, crease below the lower lip (hypertelorism) Alagille syndrome Distinct facial features, such as broad forehead, 1 in 70,000 Mutations in JAG1 gene (OMIM: 118450) pointed mandible and bulbous tip of the nose and [2] in the fingers Alfi's Syndrome Mental retardation, trigonocephaly, mongoloid 1 in 5 million Monosomy 9p or (OMIM: 158170) eyes, wide flat nasal bridge, anteverted nostrils, 9p22.2-3 deletion [3] long upper lip, cleft lip/palate, short neck, long digits mostly secondary to long middle phalanges Apert Syndrome Various manifestations of craniosynostosis with Between 1 in Mutations in FGFR2 (OMIM: 101200) cleft lip/palate. 65,000 to gene [4] 200,000 Beckwith- Characteristic facial appearance and indentations Rare Mutation or deletion of Wiedemann of the ears, a large tongue which may cause genes H19, KCNQ1OT1 Syndrome breathing, feeding or speech difficulties, one side or CDKN1C in 11p15.5 (OMIM: 130650) of the body grows more than the other chromosomal region [5- 7] Cohen Syndrome Abnormalities of the head, characteristic facial Rare Mutations in COH1 (OMIM: 216550) features including high-arched or wave-shaped gene [8] eyelids, a short philtrum, thick hair, and low hairline Cri-du-chat Abnormal larynx and epiglottis which causes a 1 in 50,000 Mutations in two Syndrome (OMIM: distinct sounding cry.