DNA-Based Identification of Spices: DNA Isolation, Whole Genome Amplification, and Polymerase Chain Reaction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Building Big Flavor
BUILDING BIG FLAVOR Watching your sodium intake? Balancing flavors is the key to a flavorful meal when reducing the salt in a dish. Think about adding these flavor enhancers instead of reaching for the salt shaker! Sweet Bitter Acidic Umami (Savory) Brings balance and Balances sweetness Brings brightness Makes a dish savory or roundness to a dish by and cuts richness - and adds a salty meaty tasting and balancing acidity and best used as flavor that enhances flavors - reach bitterness and background flavor balances for these before salt! highlighting other flavors sweetness Fruit juices, Nectars, Greens (Kale, Lemon, Lime, Tomato Products Concentrates, Chard, Dandelion, Orange and (especially canned, like Reductions, Caramelized Chicory, Watercress, Pineapple Juice, paste) Soy Sauce, Onions, Carrots, Sweet Arugula) Broccoli Vinegars, Wine, Mushrooms (especially Potatoes, Butternut Rabe, Broccoli, Tamarind, dried) Cured or brined Squash, Roasted Cabbage, Brussels Pickled Foods, foods (olives) Seaweed, Peppers, Honey, Maple Sprouts, Asparagus, Cranberries, Sour Fish Sauce, Fermented Syrup, Molasses, Dried Some Mustards, Cherries, Tomato Foods (Miso, Fermented Fruits, Tomato Paste, Grapefruit, Citrus Products Black beans, Sauerkraut) Beets, Reduced Vinegars, Rind/Zest, Beer, Aged cheeses (Parmesan, Wine, Wine, Teas (black & Blue, Gouda) Liquid Amino green) Acids, Seafood (especially dried), Worcestershire Sauce, Anchovy, Beef, Pork (especially cured), Chicken Don’t forget to read nutrition labels and watch for foods that are commonly high -

Anti–Oxidative and Anti–Inflammatory Effects of Tagetes Minuta Essential Oil in Activated Macrophages
Asian Pac J Trop Biomed 2014; 4(3): 219-227 219 Contents lists available at ScienceDirect Asian Pacific Journal of Tropical Biomedicine journal homepage: www.elsevier.com/locate/apjtb Document heading doi:10.1016/S2221-1691(14)60235-5 2014 by the Asian Pacific Journal of Tropical Biomedicine. All rights reserved. 襃 Anti-oxidative and anti-inflammatory effects of Tagetes minuta essential oil in activated macrophages 1 1 2 Parastoo Karimian , Gholamreza Kavoosi *, Zahra Amirghofran 1Biotechnology Institute, Shiraz University, Shiraz, 71441-65186, Iran 2Department of Immunology, Autoimmune Disease Research Center and Medicinal and Natural Products Chemistry Research Center, Shiraz University of Medical Sciences, Shiraz, Iran PEER REVIEW ABSTRACT Peer reviewer Objective: Tagetes minuta T. minuta To investigate antioxidant and anti-inflammatory effects of ( ) Hasan Salehi, University of Shiraz, essentialMethods: oil. T. minuta T. minuta Shiraz, Iran. In the present study essential oil was obtained from leaves of via E-mail: [email protected] hydro-distillation andT. minutathen was analyzed by gas chromatography-mass spectrometry. The anti- Comments oxidant capacity of essential oil was examined by measuring reactive oxygen,T. reactive minuta nitrogen species and hydrogen peroxide scavenging. The anti-inflammatory activity of TMO displayed an anti-oxidant essential αoil was determined through measuring NADH oxidase, inducible nitric oxide synthase property by scavenging superoxide, and TNF- mRNA expression in lipopolysacharide-stimulated murine macrophages using real- H2O2 and NO radicals, and reduced Results:time PCR. G oxidative stress. The decreased T. minutaas chromatography-mass spectrometry analysis indicated that the main components in ROS NOS (33 86%) E (19 92%) (16 15%) formation of and radicals in the β essential oil were dihydrotagetone . -

Butter Poached Prawns with Tarragon & Garlic
Butter Poached Prawns with Tarragon & Garlic the One° Precision Poacher™ With probe Butter Poached Shrimp with Tarragon & Garlic Prep 5 minutes / Cook 15 minutes the One° Precision Poacher™ Serves 2 16 large shrimp, peeled and deveined, tails intact 4 tablespoons (60g) salted butter, diced 1 tablespoon fresh tarragon, finely chopped 1 clove garlic, crushed Freshly ground black pepper, to taste Method 1. Fill the pot of the Precision Poacher with water up to the SOUS VIDE fill line. Put the egg tray into the pot. Cover with the lid and insert probe through the vent. Press METHOD button to select SOUS VIDE. Press TEMPERATURE button to select 59°C. Press TIME button to select 15 minutes. Press START to preheat water. 2. While the water is preheating, place shrimp neatly into a vacuum bag with butter cubes, tarragon, garlic and black pepper. Vacuum seal the bag. 3. When preheat has finished, the unit will beep. Drop the bag into the water, ensuring it is submerged. Cover with the lid and insert probe. 4. Press START. When cooking is complete, snip the bag and divide shrimps among two bowls. Drizzle over the garlic and tarragon butter, season. Serve with crusty bread and salad. Note: A vacuum sealer and vacuum bags are needed for this recipe. BEG800 B16 Eggs Benedict the One° Precision Poacher™ With probe Eggs Benedict Prep 10 minutes / Cook 20 minutes Serves 4 (Makes ¾ cup (200ml) hollandaise) the One° Precision Poacher™ 4 large eggs 1 tablespoon olive oil 4 portobello mushrooms 4oz (115g) shaved smoked ham 1 bunch (200g) spinach, washed and trimmed Hollandaise 3 large egg yolks 2 tablespoons lemon juice 7 tablespoons (100g) unsalted butter, cubed Salt and pepper, to season Method 1. -

21 CFR Ch. I (4–1–10 Edition) § 582.20
§ 582.20 21 CFR Ch. I (4–1–10 Edition) Common name Botanical name of plant source Marjoram, sweet .......................................................................... Majorana hortensis Moench. Mustard, black or brown .............................................................. Brassica nigra (L.) Koch. Mustard, brown ............................................................................ Brassica juncea (L.) Coss. Mustard, white or yellow .............................................................. Brassica hirta Moench. Nutmeg ........................................................................................ Myristica fragrans Houtt. Oregano (oreganum, Mexican oregano, Mexican sage, origan) Lippia spp. Paprika ......................................................................................... Capsicum annuum L. Parsley ......................................................................................... Petroselinum crispum (Mill.) Mansf. Pepper, black ............................................................................... Piper nigrum L. Pepper, cayenne ......................................................................... Capsicum frutescens L. or Capsicum annuum L. Pepper, red .................................................................................. Do. Pepper, white ............................................................................... Piper nigrum L. Peppermint .................................................................................. Mentha piperita L. Poppy seed -

Season with Herbs and Spices
Season with Herbs and Spices Meat, Fish, Poultry, and Eggs ______________________________________________________________________________________________ Beef-Allspice,basil, bay leaf, cardamon, chives, curry, Chicken or Turkey-Allspice, basil, bay leaf, cardamon, garlic, mace, marjoram, dry mustard, nutmeg, onion, cumin, curry, garlic, mace, marjoram, mushrooms, dry oregano, paprika, parsley, pepper, green peppers, sage, mustard, paprika, parsley, pepper, pineapple sauce, savory, tarragon, thyme, turmeric. rosemary, sage, savory, tarragon, thyme, turmeric. Pork-Basil, cardamom, cloves, curry, dill, garlic, mace, Fish-Bay leaf, chives, coriander, curry, dill, garlic, lemon marjoram, dry mustard, oregano, onion, parsley, pepper, juice, mace, marjoram, mushrooms, dry mustard, onion, rosemary, sage, thyme, turmeric. oregano, paprika, parsley, pepper, green peppers, sage, savory, tarragon, thyme, turmeric. Lamb-Basil, curry, dill, garlic, mace, marjoram, mint, Eggs-Basil, chili powder, chives, cumin, curry, mace, onion, oregano, parsley, pepper, rosemary, thyme, marjoram, dry mustard, onion, paprika, parsley, pepper, turmeric. green peppers, rosemary, savory, tarragon, thyme. Veal-Basil, bay leaf, curry, dill, garlic, ginger, mace, marjoram, oregano, paprika, parsley, peaches, pepper, rosemary, sage, savory, tarragon, thyme, turmeric. Vegetables Asparagus-Caraway seed, dry mustard, nutmeg, sesame Broccoli-Oregano, tarragon. seed. Cabbage-Basil, caraway seed, cinnamon,dill, mace, dry Carrots-Chili powder, cinnamon, ginger, mace, marjoram, mustard, -
Herbs Such As Spearmint
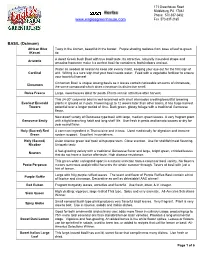
171 Greenhouse Road Middleburg, PA 17842 Phone: 570-837-0432 www.englesgreenhouse.com Fax 570-837-2165 BASIL (Ocimum) African Blue Tasty in the kitchen, beautiful in the border. Purple shading radiates from base of leaf to green (Kasar) tip. A dwarf Greek bush Basil with true basil taste. Its attractive, naturally mounded shape and Aristotle amazing fragrance make it a perfect basil for containers, both indoors and out. Water as needed all season to keep soil evenly moist, keeping your eye out for the first sign of Cardinal wilt. Wilting is a sure sign that your basil needs water. Feed with a vegetable fertilizer to ensure your bountiful harvest Cinnamon Basil is unique among basils as it leaves contain noticeable amounts of cinnamate, Cinnamon the same compound which gives cinnamon its distinctive smell. Dolce Fresca Large, sweet leaves ideal for pesto. Plants remain attractive after harvest. This 24-30” columnar basil is well-branched with short internodes creating beautiful towering Everleaf Emerald plants in ground or in pots. Flowering up to 12 weeks later than other basils, it has huge harvest Towers potential over a longer period of time. Dark green, glossy foliage with a traditional Genovese flavor. New dwarf variety of Genovese type basil with large, medium green leaves. A very fragrant plant Genovese Emily with a tight branching habit and long shelf life. Use fresh in pesto and tomato sauces or dry for year round flavor. Holy (Sacred) Red A common ingredient in Thai cuisine and in teas. Used medicinally for digestion and immune Green system support. -

Herbs Ship Week: Ship Week
Please include this first page with every order. Date: Company Name: Ordered By: Phone Number: Choice of Courier: 28904 Fraser Highway Abbotsford, BC V4Z 1G8 ': (604) 857-4944 7: (604) 857-4947 ✉: [email protected] Note: Some varieties are non-exportable # of 33 Count # of 100 # of 33 Count # of 100 Variety # of 100 URC # of 100 URC Trays Count Trays Trays Count Trays Herbs Ship Week: Ship Week: Aztec Sweet Herb Lippia dulcis Calamint Variegata N/A N/A N/A N/A N/A N/A Chamomile Flore Pleno Chamomile Treneague Cola Plant Artemisia arbotanum Curry Plant Helichrysum italicum Curry Tall Helichrysum angustifolia Gaura lindheimeri Freefolk Rosy* Glasswort Salicornia europaea Hesperozygis Midnight Mojito* Indian Mint® Satureja douglasii Jiaogulan Gynostemma pentaphyllum Lavandula allardii Meerlo* Lavandula angustifolia Artic Snow Lavandula angustifolia BeeZee™ Dark Blue Lavandula angustifolia BeeZee™ Light Blue Lavandula angustifolia BeeZee™ Pink Lavandula angustifolia BeeZee™ Power Blue Lavandula angustifolia BeeZee™ White Lavandula angustifolia Big Time Blue* Lavandula angustifolia Dwarf Blue Lavandula angustifolia Forever Blue* Lavandula angustifolia Imperial Gem Lavandula angustifolia Layla™ Blue Lavandula angustifolia Little Lady Lavandula angustifolia Melissa Lilac Lavandula angustifolia Munstead Lavandula angustifolia Munstead Hishtil's Strain Lavandula angustifolia Platinum Blonde* Lavandula angustifolia Rosea Lavandula angustifolia Thumbelina Leigh* Lavandula angustifolia Twickle Purple Lavandula angustifolia Vera Herbs 1 of 6 -

Spice Basics
SSpicepice BasicsBasics AAllspicellspice Allspice has a pleasantly warm, fragrant aroma. The name refl ects the pungent taste, which resembles a peppery compound of cloves, cinnamon and nutmeg or mace. Good with eggplant, most fruit, pumpkins and other squashes, sweet potatoes and other root vegetables. Combines well with chili, cloves, coriander, garlic, ginger, mace, mustard, pepper, rosemary and thyme. AAnisenise The aroma and taste of the seeds are sweet, licorice like, warm, and fruity, but Indian anise can have the same fragrant, sweet, licorice notes, with mild peppery undertones. The seeds are more subtly fl avored than fennel or star anise. Good with apples, chestnuts, fi gs, fi sh and seafood, nuts, pumpkin and root vegetables. Combines well with allspice, cardamom, cinnamon, cloves, cumin, fennel, garlic, nutmeg, pepper and star anise. BBasilasil Sweet basil has a complex sweet, spicy aroma with notes of clove and anise. The fl avor is warming, peppery and clove-like with underlying mint and anise tones. Essential to pesto and pistou. Good with corn, cream cheese, eggplant, eggs, lemon, mozzarella, cheese, olives, pasta, peas, pizza, potatoes, rice, tomatoes, white beans and zucchini. Combines well with capers, chives, cilantro, garlic, marjoram, oregano, mint, parsley, rosemary and thyme. BBayay LLeafeaf Bay has a sweet, balsamic aroma with notes of nutmeg and camphor and a cooling astringency. Fresh leaves are slightly bitter, but the bitterness fades if you keep them for a day or two. Fully dried leaves have a potent fl avor and are best when dried only recently. Good with beef, chestnuts, chicken, citrus fruits, fi sh, game, lamb, lentils, rice, tomatoes, white beans. -

French Tarragon in the Garden Benjamin Hudson and Dan Drost Vegetable Specialist
Revised May 2020 French Tarragon in the Garden Benjamin Hudson and Dan Drost Vegetable Specialist Summary Referred as “a chef’s best friend,” French Varieties Tarragon (Artemisia dracunculus) is an essential There are two types of tarragon: French or aromatic herb. Other common names include Russian. Russian tarragon, while not classified as estragon, dragon sagewort, or German tarragon, but being a different species, has flavor vastly inferior it should not be confused with the closely related to French tarragon. Russian tarragon is generally Russian tarragon. French tarragon may be grown as grown from seed, while French tarragon is almost an annual or as a perennial, as it is winter hardy to exclusively propagated by vegetative cuttings. zone 4. Depending on the climate, it may be Consult a reputable nursery or garden center when necessary to cover French tarragon with mulch purchasing tarragon plants to ensure you get the during the winter, when grown as a perennial. desired type. French tarragon prefers full sun, well-drained soils, grows to a height of 24 to 36 inches with a 12 to 15 How to Grow inch spread. For fresh use, harvest sprigs of French Soil: French tarragon grows best in warm, tarragon as needed or for storage harvest the entire dry, well-aerated soils and does not tolerate wet or plant and dry. saturated soils. French tarragon grows well in neutral pH soils (pH 6.5-7.5), but exhibits some preference for slightly acidic soils. Most soils in Utah are suitable for growing French tarragon provided they are well drained. Soil Preparation: Before planting, determine fertilizer needs with a soil test and then follow the fertilization recommendations given. -

Growing Herbs in Laramie County
Catherine Wissner UW Extension Service Laramie County Cheyenne, Wyoming What they offer: planting information, ancient history and lore, poetry, musings, photography, illustrations, recipes, chemical constituents and medicinal virtues of herbs. From the botanical viewpoint, an herb is a seed plant that does not produce a woody stem like a tree. But an herb will live long enough to develop flowers and seeds. Richters catalog out of Canada, list over 200 herbs, 43 different types of Basil, 40 mints, 15 Rosemary’s, 35 Sages, and 10 Beebalms. Seed Savers Exchange lists over 350 herbs. So many choices so little time….. Known as the mint family. Comprising about 210 genera and some 3,500 species. The plants are frequently aromatic in all parts include many widely used culinary, such as: Basil, Mint, Rosemary, Sage, Savory, Marjoram, Oregano, Thyme, Lavender. Mostly with opposite leaves, when crushed the foliage usually emitting various, mostly pleasant odors. Stems usually square. Flowers usually abundant and quite attractive, the sepals and corollas variously united. Calyx 2-lipped or not. Corollas strongly 2-lipped (labiate, hence the family name). Herbs fit into one or more classifications according to use - - culinary, aromatic, ornamental, and medicinal. Culinary herbs are probably the most useful to herb gardeners, having a wide range of uses in cooking. Strong herbs -- winter savory, rosemary, sage. Herbs for accent -- sweet basil, dill, mint, sweet marjoram, tarragon, thyme. Herbs for blending -- chives, parsley, summer savory.. Aromatic herbs Most have pleasant smelling flowers or foliage. Oils from aromatic herbs can also be used to produce perfumes and various scents. -

(Tagetes Minuta) Oils
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/290094383 Tagetes (Tagetes minuta) Oils Chapter · December 2016 DOI: 10.1016/B978-0-12-416641-7.00090-0 CITATIONS READS 4 3,744 2 authors, including: Wycliffe Wanzala Maasai Mara University 81 PUBLICATIONS 371 CITATIONS SEE PROFILE Some of the authors of this publication are also working on these related projects: Sustainable and Applied Health Sciences and Community Outreach/Services. View project Effects of air pollution om plants View project All content following this page was uploaded by Wycliffe Wanzala on 29 September 2017. The user has requested enhancement of the downloaded file. Author's personal copy Provided for non-commercial research and educational use only. Not for reproduction, distribution or commercial use. This chapter was originally published in the book Essential Oils in Food Preservation, Flavor and Safety. The copy attached is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research, and educational use. This includes without limitation use in instruction at your institution, distribution to specific colleagues, and providing a copy to your institution's administrator. All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution’s website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier’s permissions site at: http://www.elsevier.com/locate/permissionusematerial From Cornelius, W.W., Wycliffe, W., 2016. Tagetes (Tagetes minuta) Oils. -

Don't Pass the Salt! Low Sodium Seasoning Guide
Don’t Pass the Salt! Low Sodium Seasoning Guide 1. Resist the urge to use salt in cooking or using the salt shaker. One teaspoon of salt is equal to 2,300 mg of sodium. 2. Use spices and herbs to flavor your foods to add interest and variety. 3. Herb Blends These herb blends can be found in your local store to add flavor to the foods you enjoy. Be sure to check the label to be sure they do not contain salt or sodium on the list of ingredients. How to cook with herbs and spices: Finding “Hidden” salt on a label To release more flavor and Sodium benzoate (a aroma, finely chop fresh herbs preservative) before using in the recipe. Try using kitchen shears. Sodium nitrate (a preservative used in processed meats) Add herbs and spices at the end of the cooking time in soups and Bicarbonate of soda or baking stews. That way the flavors soda won’t cook out. Sodium pyrophosphage Add herbs and spices several hours before serving a cold dish, Monosodium glutamate such as salads and dips. A general rule of thumb with herbs and spices: one tablespoon of fresh herbs equals one teaspoon of dried herbs. Seasoning Guide Meats and Protein Suggested Seasoning Beef Allspice, basil, bay leaf, caraway seed, celery seed, chili powder, cumin, ginger, onion or garlic powder, rosemary, savory, tarragon or thyme Eggs Basil, celery seed, chili powder, curry, cumin, marjoram, rosemary and savory Fish Curry powder, dill, lemon or marjoram Lamb Curry powder, mint, onion or garlic powder or rosemary Pork Bay leaf, caraway seed, chili powder, cloves, curry powder,