Neuroblastoma Cancer Stem Cells: the Role of NXPH1 and Its Receptor Α-NRXN1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Myopia in African Americans Is Significantly Linked to Chromosome 7P15.2-14.2
Genetics Myopia in African Americans Is Significantly Linked to Chromosome 7p15.2-14.2 Claire L. Simpson,1,2,* Anthony M. Musolf,2,* Roberto Y. Cordero,1 Jennifer B. Cordero,1 Laura Portas,2 Federico Murgia,2 Deyana D. Lewis,2 Candace D. Middlebrooks,2 Elise B. Ciner,3 Joan E. Bailey-Wilson,1,† and Dwight Stambolian4,† 1Department of Genetics, Genomics and Informatics and Department of Ophthalmology, University of Tennessee Health Science Center, Memphis, Tennessee, United States 2Computational and Statistical Genomics Branch, National Human Genome Research Institute, National Institutes of Health, Baltimore, Maryland, United States 3The Pennsylvania College of Optometry at Salus University, Elkins Park, Pennsylvania, United States 4Department of Ophthalmology, University of Pennsylvania, Philadelphia, Pennsylvania, United States Correspondence: Joan E. PURPOSE. The purpose of this study was to perform genetic linkage analysis and associ- Bailey-Wilson, NIH/NHGRI, 333 ation analysis on exome genotyping from highly aggregated African American families Cassell Drive, Suite 1200, Baltimore, with nonpathogenic myopia. African Americans are a particularly understudied popula- MD 21131, USA; tion with respect to myopia. [email protected]. METHODS. One hundred six African American families from the Philadelphia area with a CLS and AMM contributed equally to family history of myopia were genotyped using an Illumina ExomePlus array and merged this work and should be considered co-first authors. with previous microsatellite data. Myopia was initially measured in mean spherical equiv- JEB-W and DS contributed equally alent (MSE) and converted to a binary phenotype where individuals were identified as to this work and should be affected, unaffected, or unknown. -

Genetic Profiling of a Cone-Dominated Retina and Cone Photoreceptor Subtypes
Genetic profiling of a cone-dominated retina and cone photoreceptor subtypes Vincent P. Kunze Date of birth June 8, 1986 Place of birth Kempten (Allgäu) Accepted thesis Doctor rerum naturalium (Dr. rer. nat.) Institute for Biology and Environmental Sciences Carl von Ossietzky Universität Oldenburg First examiner: Prof. Dr. Karl-Wilhelm Koch Second examiner: apl. Prof. Dr. Karin Dedek Date of thesis defense: December 18, 2017 2 Abstract Abstract Mammals have two major types of sensory neurons in the retina: rods, specialized for vision in dim-light, and cones for vision in well-lit conditions and the perception of color. In my thesis, I characterized features of cone photoreceptor subtypes and the cone-dominated retinae of the tree shrew and the thirteen-lined ground squirrel. In tree shrew, I described morphological and transcriptomic changes during development. Compared to mice, the general morphology of the developing retina was similar. There were subtle differences in the onset of transcription factors and in the relative timepoints of photoreceptor genesis. The transcriptomic analysis revealed and onset Wif1-expression after birth, a gene that could potentially suppress rod development in the tree shrew. Furthermore, I looked at molecular differences in cone photoreceptor subtypes. Most mammals have two cone types, namely S- and M-cones. They diverge in their sensitivity to different wavelengths of light, based on their expression of different light-sensitive proteins: S-opsin for blue light and M-opsin for green light. Until now, cones have been classified mostly by the opsin they express. The purpose of this project was to identify genetic differences in cones and, more specifically, to find genes that are involved in cone synapse formation. -

Alzheimer's Disease Neuroimaging Initiative Biomarkers As Quantitative
Alzheimer’s & Dementia 6 (2010) 265–273 Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: Genetics core aims, progress, and plans Andrew J. Saykina,b,*, Li Shena,c, Tatiana M. Foroudb, Steven G. Potkind, Shanker Swaminathana,b, Sungeun Kima,c, Shannon L. Risachera, Kwangsik Nhoa,e, Matthew J. Huentelmanf, David W. Craigf, Paul M. Thompsong, Jason L. Steing, Jason H. Mooreh,i, Lindsay A. Farrerj, Robert C. Greenj, Lars Bertramk, Clifford R. Jack, Jr.l, Michael W. Weinerm,n,o,p; and the Alzheimer’s Disease Neuroimaging Initiative aDepartment of Radiology and Imaging Sciences, Center for Neuroimaging, Indiana University School of Medicine, Indianapolis, IN, USA bDepartment of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, USA cCenter for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN, USA dDepartment of Psychiatry and Human Behavior, University of California, Irvine, CA, USA eDivision of Medical Informatics, Regenstrief Institute, Indianapolis, IN, USA fNeurogenomics Division, The Translational Genomics Research Institute, Phoenix, AZ, USA gLaboratory of Neuro Imaging, UCLA School of Medicine, Los Angeles, CA, USA hDepartment of Genetics, Computational Genetics Laboratory, Dartmouth Medical School, Lebanon, NH, USA iDepartment of Community and Family Medicine, Computational Genetics Laboratory, Dartmouth Medical School, Lebanon, NH, USA jDepartments of Medicine (Genetics Program), Neurology, Epidemiology, Genetics, -

Cisplatin Treatment of Testicular Cancer Patients Introduces Long-Term Changes in the Epigenome Cecilie Bucher-Johannessen1, Christian M
Bucher-Johannessen et al. Clinical Epigenetics (2019) 11:179 https://doi.org/10.1186/s13148-019-0764-4 RESEARCH Open Access Cisplatin treatment of testicular cancer patients introduces long-term changes in the epigenome Cecilie Bucher-Johannessen1, Christian M. Page2,3, Trine B. Haugen4 , Marcin W. Wojewodzic1, Sophie D. Fosså1,5,6, Tom Grotmol1, Hege S. Haugnes7,8† and Trine B. Rounge1,9*† Abstract Background: Cisplatin-based chemotherapy (CBCT) is part of standard treatment of several cancers. In testicular cancer (TC) survivors, an increased risk of developing metabolic syndrome (MetS) is observed. In this epigenome- wide association study, we investigated if CBCT relates to epigenetic changes (DNA methylation) and if epigenetic changes render individuals susceptible for developing MetS later in life. We analyzed methylation profiles, using the MethylationEPIC BeadChip, in samples collected ~ 16 years after treatment from 279 Norwegian TC survivors with known MetS status. Among the CBCT treated (n = 176) and non-treated (n = 103), 61 and 34 developed MetS, respectively. We used two linear regression models to identify if (i) CBCT results in epigenetic changes and (ii) epigenetic changes play a role in development of MetS. Then we investigated if these changes in (i) and (ii) links to genes, functional networks, and pathways related to MetS symptoms. Results: We identified 35 sites that were differentially methylated when comparing CBCT treated and untreated TC survivors. The PTK6–RAS–MAPk pathway was significantly enriched with these sites and infers a gene network of 13 genes with CACNA1D (involved in insulin release) as a network hub. We found nominal MetS-associations and a functional gene network with ABCG1 and NCF2 as network hubs. -

NIH Public Access Author Manuscript Alzheimers Dement
NIH Public Access Author Manuscript Alzheimers Dement. Author manuscript; available in PMC 2010 May 12. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Alzheimers Dement. 2010 May ; 6(3): 265±273. doi:10.1016/j.jalz.2010.03.013. Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: Genetics core aims, progress, and plans Andrew J. Saykina,b,*, Li Shena,c, Tatiana M. Foroudb, Steven G. Potkind, Shanker Swaminathana,b, Sungeun Kima,c, Shannon L. Risachera, Kwangsik Nhoa,e, Matthew J. Huentelmanf, David W. Craigf, Paul M. Thompsong, Jason L. Steing, Jason H. Mooreh,i, Lindsay A. Farrerj, Robert C. Greenj, Lars Bertramk, Clifford R. Jack Jr.l, Michael W. Weinerm,n,o,p, and the Alzheimer’s Disease Neuroimaging Initiative aDepartment of Radiology and Imaging Sciences, Center for Neuroimaging, Indiana University School of Medicine, Indianapolis, IN, USA bDepartment of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, USA cCenter for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN, USA dDepartment of Psychiatry and Human Behavior, University of California, Irvine, CA, USA eDivision of Medical Informatics, Regenstrief Institute, Indianapolis, IN, USA fNeurogenomics Division, The Translational Genomics Research Institute, Phoenix, AZ, USA gLaboratory of Neuro Imaging, UCLA School of Medicine, Los Angeles, CA, USA hDepartment of Genetics, Computational Genetics Laboratory, -
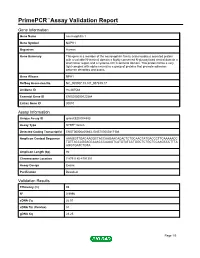
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name neurexophilin 1 Gene Symbol NXPH1 Organism Human Gene Summary This gene is a member of the neurexophilin family and encodes a secreted protein with a variable N-terminal domain a highly conserved N-glycosylated central domain a short linker region and a cysteine-rich C-terminal domain. This protein forms a very tight complex with alpha neurexins a group of proteins that promote adhesion between dendrites and axons. Gene Aliases NPH1 RefSeq Accession No. NC_000007.13, NT_007819.17 UniGene ID Hs.487564 Ensembl Gene ID ENSG00000122584 Entrez Gene ID 30010 Assay Information Unique Assay ID qHsaCED0004583 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000405863, ENST00000417186 Amplicon Context Sequence AAAGGTTGACAAGGCTACCAAGAACACACTCTGCAACTATGACCCTTCAAAAACC TGTTACCAGGAGCAAACCCAAAGTCATGTATCCTGGCTCTGCTCCAAGCCCTTTA AGGTGATCTGTA Amplicon Length (bp) 92 Chromosome Location 7:8791180-8791301 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 98 R2 0.9996 cDNA Cq 26.91 cDNA Tm (Celsius) 81 gDNA Cq 23.25 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report NXPH1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 3/5 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Molecular Sciences High-Resolution Chromosome Ideogram Representation of Currently Recognized Genes for Autism Spectrum Disorder
Int. J. Mol. Sci. 2015, 16, 6464-6495; doi:10.3390/ijms16036464 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article High-Resolution Chromosome Ideogram Representation of Currently Recognized Genes for Autism Spectrum Disorders Merlin G. Butler *, Syed K. Rafi † and Ann M. Manzardo † Departments of Psychiatry & Behavioral Sciences and Pediatrics, University of Kansas Medical Center, Kansas City, KS 66160, USA; E-Mails: [email protected] (S.K.R.); [email protected] (A.M.M.) † These authors contributed to this work equally. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-913-588-1873; Fax: +1-913-588-1305. Academic Editor: William Chi-shing Cho Received: 23 January 2015 / Accepted: 16 March 2015 / Published: 20 March 2015 Abstract: Recently, autism-related research has focused on the identification of various genes and disturbed pathways causing the genetically heterogeneous group of autism spectrum disorders (ASD). The list of autism-related genes has significantly increased due to better awareness with advances in genetic technology and expanding searchable genomic databases. We compiled a master list of known and clinically relevant autism spectrum disorder genes identified with supporting evidence from peer-reviewed medical literature sources by searching key words related to autism and genetics and from authoritative autism-related public access websites, such as the Simons Foundation Autism Research Institute autism genomic database dedicated to gene discovery and characterization. Our list consists of 792 genes arranged in alphabetical order in tabular form with gene symbols placed on high-resolution human chromosome ideograms, thereby enabling clinical and laboratory geneticists and genetic counsellors to access convenient visual images of the location and distribution of ASD genes. -
Exploring Differentially Methylated Genes in Vulvar Squamous Cell Carcinoma
cancers Article Exploring Differentially Methylated Genes in Vulvar Squamous Cell Carcinoma Shatavisha Dasgupta 1,* , Patricia C. Ewing-Graham 1, Sigrid M. A. Swagemakers 1,2, Thierry P. P. van den Bosch 1 , Peggy N. Atmodimedjo 1, Michael M. P. J. Verbiest 3, Marit de Haan 1, Helena C. van Doorn 4, Peter J. van der Spek 1,2, Senada Koljenovi´c 1 and Folkert J. van Kemenade 1 1 Department of Pathology, Erasmus MC, University Medical Centre Rotterdam, 3000 CA Rotterdam, The Netherlands; [email protected] (P.C.E.-G.); [email protected] (S.M.A.S.); [email protected] (T.P.P.v.d.B.); [email protected] (P.N.A.); [email protected] (M.d.H.); [email protected] (P.J.v.d.S.); [email protected] (S.K.); [email protected] (F.J.v.K.) 2 Department of Clinical Bioinformatics, Erasmus MC, University Medical Centre Rotterdam, 3000 CA Rotterdam, The Netherlands 3 Department of Internal Medicine, Erasmus MC, University Medical Centre Rotterdam, 3000 CA Rotterdam, The Netherlands; [email protected] 4 Department of Gynecologic Oncology, Erasmus MC Cancer Institute, University Medical Centre Rotterdam, 3000 CA Rotterdam, The Netherlands; [email protected] * Correspondence: [email protected] Citation: Dasgupta, S.; Simple Summary: Vulvar squamous cell carcinoma (VSCC) is the most common form of vulvar Ewing-Graham, P.C.; Swagemakers, S.M.A.; van den Bosch, T.P.P.; malignancy, and its incidence has increased in recent years. For better diagnosis and prognostication, Atmodimedjo, P.N.; Verbiest, M.M.P.J.; and to expand available treatment options, molecular characterization of VSCC is crucial. -
7P21.3 Together with a 12P13.32 Deletion in a Patient with Microcephaly—Does 12P13.32 Locus Possibly Comprises a Candidate Gene Region for Microcephaly?
7p21.3 Together With a 12p13.32 Deletion in a Patient With Microcephaly—Does 12p13.32 Locus Possibly Comprises a Candidate Gene Region for Microcephaly? Rinčić, Martina; Radoš, Milan; Kopić, Janja; Krsnik, Željka; Liehr, Thomas Source / Izvornik: Frontiers in Molecular Neuroscience, 2021, 14 Journal article, Published version Rad u časopisu, Objavljena verzija rada (izdavačev PDF) https://doi.org/10.3389/fnmol.2021.613091 Permanent link / Trajna poveznica: https://urn.nsk.hr/urn:nbn:hr:105:079645 Rights / Prava: Attribution 4.0 International Download date / Datum preuzimanja: 2021-09-27 Repository / Repozitorij: Dr Med - University of Zagreb School of Medicine Digital Repository DATA REPORT published: 04 February 2021 doi: 10.3389/fnmol.2021.613091 7p21.3 Together With a 12p13.32 Deletion in a Patient With Microcephaly—Does 12p13.32 Locus Possibly Comprises a Candidate Gene Region for Microcephaly? Martina Rincic 1*, Milan Rados 1, Janja Kopic 1, Zeljka Krsnik 1 and Thomas Liehr 2 1 Croatian Institute for Brain Research, School of Medicine, University of Zagreb, Zagreb, Croatia, 2 Jena University Hospital, Friedrich Schiller University, Institute of Human Genetics, Jena, Germany Keywords: microcephaly, magnetic resonace imaging (MRI), intellectual disability—genetics∗, PRMT8, array comparative genetic hybridization (aCGH) INTRODUCTION Genomic regions of copy number changes, gains and losses intermediate in size named copy number variant (CNV) are not only important source of genomic variation in the general population, but are also well-known causes of many neurodevelopmental disorders (Lee and Scherer, 2010; Weise et al., 2012). CNVs can have impact on gene/genes function via several different mechanism (Hehir-Kwa et al., 2013). -

Breast Carcinoma with Amplified HER2: a Gene Expression Signature Specific for Trastuzumab Resistance and Poor Prognosis
Modern Pathology (2010) 23, 1364–1378 1364 & 2010 USCAP, Inc. All rights reserved 0893-3952/10 $32.00 Breast carcinoma with amplified HER2: a gene expression signature specific for trastuzumab resistance and poor prognosis Thaer Khoury1, Kazunori Kanehira1, Dan Wang2, Foluso Ademuyiwa3, Wilfrido Mojica4, Richard Cheney1, Carl Morrison1, Jeffrey Conroy5, Norma Nowak5 and Song Liu2 1Department of Pathology, Roswell Park Cancer Institute, Buffalo, NY, USA; 2Department of Biostatistics, Roswell Park Cancer Institute, Buffalo, NY, USA; 3Department of Medicine, Roswell Park Cancer Institute, Buffalo, NY, USA; 4Department of Pathology, Roswell Park Cancer Institute, University at Buffalo, Buffalo, NY, USA and 5Department of Microarray Facility, Cancer Genetics, Roswell Park Cancer Institute, Buffalo, NY, USA Recent trials have shown remarkable efficacy from combined trastuzumab and chemotherapy in the adjuvant setting of breast cancer. In spite of these successes, refractory breast cancer has emerged as a clinically problematic outcome for a subset of patients managed this way. In an effort to clarify and optimize the treatment regimens for breast cancer patients who are candidates to receive trastuzumab, we sought to analyze whether a distinctive genetic signature could be characterized that would reliably predict the treatment outcome. The ability to predict who will respond and who will become refractory to this agent will allow for improved, rational clinical management of these patients and further stratify the personalized nature of this treatment regimen. In this study, 41 consecutive cases of breast carcinoma with well-documented amplification of the human epidermal growth factor receptor-2 gene and corresponding banked fresh-frozen tissue were identified and divided into two separate groups based on whether they received trastuzumab or not. -

Neurexin 1 Variants As Risk Factors for Suicide Death
Molecular Psychiatry www.nature.com/mp ARTICLE OPEN Neurexin 1 variants as risk factors for suicide death Nancy William 1, Carsten Reissner2, Robert Sargent3, Todd M. Darlington4, Emily DiBlasi1, Qingqin S. Li 5, Brooks Keeshin6, 7 8 1 3 7 1 9 William B. Callor , Elliott Ferris , Leslie Jerominski , Ken R.✉ Smith , Erik D. Christensen , Douglas M. Gray , Nicola J. Camp , Markus Missler 2, Megan E. Williams8 and Hilary Coon1 © The Author(s) 2021 Suicide is a significant public health concern with complex etiology. Although the genetic component of suicide is well established, the scope of gene networks and biological mechanisms underlying suicide has yet to be defined. Previously, we reported genome- wide evidence that neurexin 1 (NRXN1), a key synapse organizing molecule, is associated with familial suicide risk. Here we present new evidence for two non-synonymous variants (rs78540316; P469S and rs199784139; H885Y) associated with increased familial risk of suicide death. We tested the impact of these variants on binding interactions with known partners and assessed functionality in a hemi-synapse formation assay. Although the formation of hemi-synapses was not altered with the P469S variant relative to wild- type, both variants increased binding to the postsynaptic binding partner, leucine-rich repeat transmembrane neuronal 2 (LRRTM2) in vitro. Our findings indicate that variants in NRXN1 and related synaptic genes warrant further study as risk factors for suicide death. Molecular Psychiatry; https://doi.org/10.1038/s41380-021-01190-2 INTRODUCTION of interest that allow us to pursue targeted follow-up of rare Suicide is the 10th leading cause of death in the U.S and accounts functional variants in an additional large, population-ascertained for almost 800,000 deaths per year globally [1]. -

Structure-Function Relationships of Rna and Protein in Synaptic Plasticity
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Structure-Function Relationships Of Rna And Protein In Synaptic Plasticity Sarah Middleton University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, Biology Commons, and the Neuroscience and Neurobiology Commons Recommended Citation Middleton, Sarah, "Structure-Function Relationships Of Rna And Protein In Synaptic Plasticity" (2017). Publicly Accessible Penn Dissertations. 2474. https://repository.upenn.edu/edissertations/2474 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2474 For more information, please contact [email protected]. Structure-Function Relationships Of Rna And Protein In Synaptic Plasticity Abstract Structure is widely acknowledged to be important for the function of ribonucleic acids (RNAs) and proteins. However, due to the relative accessibility of sequence information compared to structure information, most large genomics studies currently use only sequence-based annotation tools to analyze the function of expressed molecules. In this thesis, I introduce two novel computational methods for genome-scale structure-function analysis and demonstrate their application to identifying RNA and protein structures involved in synaptic plasticity and potentiation—important neuronal processes that are thought to form the basis of learning and memory. First, I describe a new method for de novo identification of RNA secondary structure motifs enriched in co-regulated transcripts. I show that this method can accurately identify secondary structure motifs that recur across three or more transcripts in the input set with an average recall of 0.80 and precision of 0.98. Second, I describe a tool for predicting protein structural fold from amino acid sequence, which achieves greater than 96% accuracy on benchmarks and can be used to predict protein function and identify new structural folds.