On Parameters of the Human Genome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Linkage and Genetic Mapping 4TH PAGES © Jones & Bartlett Learning, LLC
© Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION Gene Linkage and © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC 4NOTGenetic FOR SALE OR DISTRIBUTIONMapping NOT FOR SALE OR DISTRIBUTION CHAPTER ORGANIZATION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR4.1 SALELinked OR alleles DISTRIBUTION tend to stay 4.4NOT Polymorphic FOR SALE DNA ORsequences DISTRIBUTION are together in meiosis. 112 used in human genetic mapping. 128 The degree of linkage is measured by the Single-nucleotide polymorphisms (SNPs) frequency of recombination. 113 are abundant in the human genome. 129 The frequency of recombination is the same SNPs in restriction sites yield restriction for coupling and repulsion heterozygotes. 114 fragment length polymorphisms (RFLPs). 130 © Jones & Bartlett Learning,The frequency LLC of recombination differs © Jones & BartlettSimple-sequence Learning, repeats LLC (SSRs) often NOT FOR SALE OR DISTRIBUTIONfrom one gene pair to the next. NOT114 FOR SALEdiffer OR in copyDISTRIBUTION number. 131 Recombination does not occur in Gene dosage can differ owing to copy- Drosophila males. 115 number variation (CNV). 133 4.2 Recombination results from Copy-number variation has helped human populations adapt to a high-starch diet. 134 crossing-over between linked© Jones alleles. & Bartlett Learning,116 LLC 4.5 Tetrads contain© Jonesall & Bartlett Learning, LLC four products of meiosis. -

Integrating Genetic Linkage Maps with Pachytene Chromosome Structure in Maize
Copyright 2004 by the Genetics Society of America Integrating Genetic Linkage Maps With Pachytene Chromosome Structure in Maize Lorinda K. Anderson,*,1 Naser Salameh,† Hank W. Bass,‡ Lisa C. Harper,§ W. Z. Cande,§ Gerd Weber† and Stephen M. Stack* *Department of Biology, Colorado State University, Fort Collins, Colorado 80523, †Department of Plant Breeding and Biotechnology, University of Hohenheim, D-70593 Stuttgart, Germany, ‡Department of Biological Science, Florida State University, Tallahassee, Florida 32306 and §Department of Molecular and Cell Biology, University of California, Berkeley, California 94720 Manuscript received November 4, 2003 Accepted for publication January 9, 2004 ABSTRACT Genetic linkage maps reveal the order of markers based on the frequency of recombination between markers during meiosis. Because the rate of recombination varies along chromosomes, it has been difficult to relate linkage maps to chromosome structure. Here we use cytological maps of crossing over based on recombination nodules (RNs) to predict the physical position of genetic markers on each of the 10 chromosomes of maize. This is possible because (1) all 10 maize chromosomes can be individually identified from spreads of synaptonemal complexes, (2) each RN corresponds to one crossover, and (3) the frequency of RNs on defined chromosomal segments can be converted to centimorgan values. We tested our predic- tions for chromosome 9 using seven genetically mapped, single-copy markers that were independently mapped on pachytene chromosomes using in situ hybridization. The correlation between predicted and observed locations was very strong (r2 ϭ 0.996), indicating a virtual 1:1 correspondence. Thus, this new, high-resolution, cytogenetic map enables one to predict the chromosomal location of any genetically mapped marker in maize with a high degree of accuracy. -

Primer on Molecular Genetics
DOE Human Genome Program Primer on Molecular Genetics Date Published: June 1992 U.S. Department of Energy Office of Energy Research Office of Health and Environmental Research Washington, DC 20585 The "Primer on Molecular Genetics" is taken from the June 1992 DOE Human Genome 1991-92 Program Report. The primer is intended to be an introduction to basic principles of molecular genetics pertaining to the genome project. Human Genome Management Information System Oak Ridge National Laboratory 1060 Commerce Park Oak Ridge, TN 37830 Voice: 865/576-6669 Fax: 865/574-9888 E-mail: [email protected] 2 Contents Primer on Molecular Introduction ............................................................................................................. 5 Genetics DNA............................................................................................................................... 6 Genes............................................................................................................................ 7 Revised and expanded Chromosomes ............................................................................................................... 8 by Denise Casey (HGMIS) from the Mapping and Sequencing the Human Genome ...................................... 10 primer contributed by Charles Cantor and Mapping Strategies ..................................................................................................... 11 Sylvia Spengler Genetic Linkage Maps ........................................................................................... -

Genetic Markers, Map Construction, and Their Application in Plant Breeding Jack E
Genetic Markers, Map Construction, and Their Application in Plant Breeding Jack E. Staub1 and Felix C. Serquen2 Vegetable Crops Research, U. S. Department of Agriculture, Agricultural Research Service, Department of Horticulture, University of Wisconsin–Madison, WI 53706 Manju Gupta3 Mycogen Plant Sciences, Madison Laboratories, 5649 East Buckeye Road, Madison, WI 53716 The genetic improvement of a species in a bewildering array of new terms. For scien- RFLPs. Restriction fragment length poly- through artificial selection depends on the tists who have a peripheral interest in genome morphisms (RFLPs) are detected by the use of ability to capitalize on genetic effects that can mapping, but would like to understand the restriction enzymes that cut genomic DNA be distinguished from environmental effects. potential role of MAS in plant improvement, molecules at specific nucleotide sequences Phenotypic selection based on traits that are the wealth of information currently being pro- (restriction sites), thereby yielding variable- conditioned by additive allelic effects can pro- duced in this area can lead to considerable size DNA fragments (Fig. 1). Identification of duce dramatic, economically important confusion. The purpose of this paper is to genomic DNA fragments is made by Southern changes in breeding populations. Genetic describe available marker types and examine blotting, a procedure whereby DNA fragments, markers—heritable entities that are associated factors critical for their use in map construc- separated by electrophoresis, are transferred with economically important traits—can be tion and MAS. This review clarifies how ge- to nitrocellulose or nylon filter (Southern, used by plant breeders as selection tools netic markers are used in map construction 1975). -

Uncovering Cryptic Asexuality in Daphnia Magna by RAD Sequencing
GENETICS | INVESTIGATION Uncovering Cryptic Asexuality in Daphnia magna by RAD Sequencing Nils Svendsen,*,1 Celine M. O. Reisser,*,†,1 Marinela Dukic,´ ‡ Virginie Thuillier,† Adeline Ségard,* Cathy Liautard-Haag,† Dominique Fasel,† Evelin Hürlimann,† Thomas Lenormand,* Yan Galimov,§ and Christoph R. Haag*,†,2 *Centre d’Ecologie Fonctionnelle et Evolutive (CEFE)–Unité Mixte de Recherche 5175, Centre National de la Recherche Scientifique (CNRS)–Université de Montpellier–Université Paul-Valéry Montpellier–Ecole Pratique des Hautes Etudes (EPHE), campus CNRS, 19, 34293 Montpellier Cedex 5, France, †Ecology and Evolution, University of Fribourg, 1700 Fribourg, Switzerland, ‡Zoology Institute, Evolutionary Biology, University of Basel, 4051 Basel, Switzerland, and §Koltsov Institute of Developmental Biology, Russian Academy of Sciences, 119334 Moscow, Russia ABSTRACT The breeding systems of many organisms are cryptic and difficult to investigate with observational data, yet they have profound effects on a species’ ecology, evolution, and genome organization. Genomic approaches offer a novel, indirect way to investigate breeding systems, specifically by studying the transmission of genetic information from parents to offspring. Here we exemplify this method through an assessment of self-fertilization vs. automictic parthenogenesis in Daphnia magna. Self-fertilization reduces heterozygosity by 50% compared to the parents, but under automixis, whereby two haploid products from a single meiosis fuse, the expected heterozygosity reduction depends on -
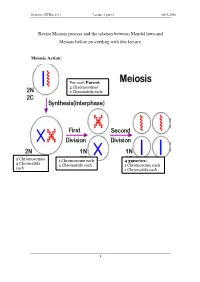
Revise Meiosis Process and the Relation Between Mendel Laws and Meiosis Before Proceeding with This Lecture
Genetics (BTBio 211) Lecture 3 part 2 2015-2016 Revise Meiosis process and the relation between Mendel laws and Meiosis before proceeding with this lecture. Meiosis Action: For each Parent 2 Chromosomes 2 Chromatids each 2 Chromosomes 1 Chromosome each 4 gametes: 4 Chromatid s 4 Chromatids each 1 Chromosome each each 1 Chromatids each 1 Genetics (BTBio 211) Lecture 3 part 2 2015-2016 LINKAGE AND CHROMOSOME MAPPING IN EUKARYOTES I. LINKAGE Genetic linkage is the tendency of genes that are located proximal to each other on a chromosome to be inherited together during meiosis. Genes whose loci are nearer to each other are less likely to be separated onto different chromatids during chromosomal crossover, and are therefore said to be genetically linked. Linked genes: Genes that are inherited together with other gene(s) in form of single unit as they are located on the same chromosome. For example: in fruit flies the genes for eye color and the genes for wing length are on the same chromosome, thus are inherited together. A couple of genes on chromosomes may be present either on the different or on the same chromosome. 1. The independent assortment of two genes located on different chromosomes. Mendel’s Law of Independent Assortment: during gamete formation, segregation of one gene pair is independent of other gene pairs because the traits he studied were determined by genes on different chromosomes. Consider two genes A and B, each with two alleles A a and B b on separate (different) chromosomes. 2 Genetics (BTBio 211) Lecture 3 part 2 2015-2016 Gametes of non-homologous chromosomes assort independently at anaphase producing 4 different genotypes AB, ab, Ab and aB with a genotypic ratio 1:1:1:1. -

Meiosis, Recombination, and Interference
Meiosis, recombination, and interference Karl W Broman Department of Biostatistics Johns Hopkins University Baltimore, Maryland, USA www.biostat.jhsph.edu/˜kbroman Outline Mitosis and meiosis Chiasmata, crossovers Genetic distance Genetic markers, recombination Chromatid and chiasma interference Mather’s formula The count-location model ¢ The gamma model and the ¡ model Data: humans and mice Mitosis: ordinary cell division Chromosomes Chr's pull apart duplicate and cell divides Chromosomes line up Meiosis: production of sex cells Chr's pull apart and cells divide Chr's exchange material and cell divides Chromosomes duplicate Chromosomes pair up The exchange process Vocabulary Four-strand bundle Meiotic products Sister chromatids Non-sister chromatids Chiasma, chiasmata Crossovers Obligate chiasma Genetic distance Two points are d Morgans apart if the average number of crossovers per meiotic product in the intervening interval is d. Usual units: centiMorgan (cM); 100 cM = 1 Morgan ¡ Genetic distance Physical distance The intensity of the crossover process varies by Sex Individual Chromosome Position on chromosome Temperature But we don’t observe crossovers Crossover Crossovers generally not process observeable Marker We instead observe the origin data of DNA at marker loci. odd no. crossovers = recombination event even no. crossovers = no recombination Recombination fraction = Pr(recombination event in interval) Microsatellite markers aka Short Tandem Repeat Polymorphisms (STRPs) GA T AGA T A GA T A CT A TCT A T CT A T Tandem repeat of something like GATA at a specific position in the genome. − Number of repeats varies Use PCR to “amplify” region Use gel electrophoresis to determine length of region + Map functions Connect genetic distance (average no. -

Genetic^And Physical Mapping Studies on Mouse Chromosome 2
Genetic^and Physical Mapping Studies on Mouse Chromosome 2 by Stavros Males A thesis submitted for the degree of Doctor of Philosophy at the University of London May 1995 The Galton Laboratory Department of Genetics and Biometry University College London ProQuest Number: 10018553 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest. ProQuest 10018553 Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 Abstract This thesis describes two genetic maps of mouse chromosome 2 (MMU2), genetic maps relative to the wasted {wsf) and Ragged (Ra) mutations on distal MMU2 and a physical map of the region likely to contain the latter two genes. The first two maps include 18 new PCR markers which were isolated from a subgenomic library constructed from a mouse-hamster somatic cell hybrid line whose main mouse-genome component was MMU2. Fourteen of these loci define microsatellite sequences and four represent randomly chosen DNA sequences. The maps were constructed using two interspecific backcrosses established using a laboratory mouse strain and mice from the related species Mus spretus. The inheritance pattern on MMU2 reveals loci that exhibit significant segregation distortion (SD) in males. -

Conceptualizing Kinship and Dependence on Centimorgan
Volume-03 ISSN: 2455-3085 (Online) Issue-12 RESEARCH REVIEW International Journal of Multidisciplinary December -2018 www.rrjournals.com [UGC Listed Journal] Conceptualizing Kinship and Dependence on Centimorgan *1Archisman Roy and 2Tushar Banerjee *1Student (Author), School of Science, AOSHS, Asansol (India) 2Research Scholar, University, Department of Zoology, SSSUTMS, Bhopal (India) ARTICLE DETAILS ABSTRACT Article History Genetics has been one of the most interesting and wildly applauded subject areas among Published Online: 10 December 2018 biologists. Precisely this subject deal with genes and the prime purpose of this biological element is to create new sanguine relations. Our paper encounters an intriguing way of Keywords analyzing kinship and its origin in genetics. This piece of research firstly goes through Genes, Chromosomes, loci, Morgan, introducing the whole context in detail with the right citations and refereeing validated modeling, homologous. researches, followingly it pacifies the sources of data with enough lucidity. The main text contains conceptualizing the whole subject matter quite engagingly as well as ponders over *Corresponding Author a detailed description of how kinship depends on centimorgans and loci of different alleles. Email:archismanroy2002[at]gmail.com Next to that overall summary of the previous elucidations have been assorted to precede a precise and qualitative analysis of the whole report. At last, the write-up ends with the complete texture of lucidity and contents relating to sanguine kinship and its dependence on the position of genes with centimorgans. 1. Introduction genes already existing thereon chromosome, the It is obvious to have so many confusions within the minds method is understood as “recombination”. of the many enthusiastic genealogists on precisely what a III. -

Principles, Requirements and Prospects of Genetic Mapping in Plants
African Journal of Biotechnology Vol. 5 (25), pp. 2569-2587, 29 December 2006 Available online at http://www.academicjournals.org/AJB ISSN 1684–5315 © 2006 Academic Journals Review Principles, requirements and prospects of genetic mapping in plants K. Semagn1*, Å. Bjørnstad2 and M. N. Ndjiondjop1 1Africa Rice Center (WARDA), 01 BP 2031, Cotonou, Benin. 2Norwegian University of Life Sciences, Department of Plant and Environmental Sciences, P.O. Box 5003, N-1432, Ås, Norway Accepted 24 November, 2006 Genetic mapping (also known as linkage mapping or meiotic mapping) refers to the determination of the relative position and distances between markers along chromosomes. Genetic map distances between two markers are defined as the mean number of recombination events, involving a given chromatid, in that region per meiosis. Genetic map construction requires that the researcher develop appropriate mapping population, decide the sample size and type of molecular marker(s) for genotyping, genotype the mapping population with sufficient number of markers, and perform linkage analyses using statistical programs. The construction of detailed genetic maps with high levels of genome coverage is a first step for localizing genes or quantitative trait loci (QTL) that are associated with economically important traits, marker assisted selection, comparative mapping between different species, a framework for anchoring physical maps, and the basis for map-based cloning of genes. Highly reproducible, high throughput, codominant, and transferable molecular markers, especially developed from expressed regions, are sought to increase the utility of genetic maps. This article reviews the principles, requirements, and future prospects of genetic mapping in plants. Key words: Crop improvement, JoinMap, linkage mapping, meiotic mapping, molecular markers, physical map. -

Glossary of Biotechnology and Genetic Engineering 1
FAO Glossary of RESEARCH AND biotechnology TECHNOLOGY and PAPER genetic engineering 7 A. Zaid H.G. Hughes E. Porceddu F. Nicholas Food and Agriculture Organization of the United Nations Rome, 1999 – ii – The designations employed and the presentation of the material in this document do not imply the expression of any opinion whatsoever on the part of the United Nations or the Food and Agriculture Organization of the United Nations concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. ISBN: 92-5-104369-8 ISSN: 1020-0541 All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying or otherwise, without the prior permission of the copyright owner. Applications for such permission, with a statement of the purpose and extent of the reproduction, should be addressed to the Director, Information Division, Food and Agriculture Organization of the United Nations, Viale delle Terme di Caracalla, 00100 Rome, Italy. © FAO 1999 – iii – PREFACE Biotechnology is a general term used about a very broad field of study. According to the Convention on Biological Diversity, biotechnology means: “any technological application that uses biological systems, living organisms, or derivatives thereof, to make or modify products or processes for specific use.” Interpreted in this broad sense, the definition covers many of the tools and techniques that are commonplace today in agriculture and food production. If interpreted in a narrow sense to consider only the “new” DNA, molecular biology and reproductive technology, the definition covers a range of different technologies, including gene manipulation, gene transfer, DNA typing and cloning of mammals. -

Discovery of the Sex-Linked Traits. in 1910, Morgan Published Details of His Research in an Article Titled “Sex Limited Inheritance in Drosophila"
Lecture 4. Chromosome theory of inheritance. Sex determination and the inheritance of sex-linked traits. 1. Chromosome theory of inheritance. 2. Chromosome mapping. 3. Sex determination and the inheritance of sex-linked traits. In 1865 Gregor Mendel has discovered laws of heredity. But he came to the conclusion of the heredity principles without the knowledge of genes and chromosomes. Over the next decades after Mendel’s findings cytological bases of heredity become more understandable. In 1869 Johannes Friedrich Miescher was the first researcher to isolate and identify nucleic acid in pus cells. In 1878 Walther Flemming described chromatin threads in cell nucleus and behavior of chromosomes during mitosis. In 1890 August Weismann described role of meiosis for reproduction and heredity. In 1888 Heinrich Waldeyer has coined the term “chromosome” to describe basophilic stained filaments inside the cell nucleus. The speculation that chromosomes might be the key to understanding heredity led several scientists to examine Mendel’s publications and re- evaluate his model in terms of the behavior of chromosomes during mitosis and meiosis. In 1902, Theodor Boveri (right) observed that proper embryonic development of sea urchins does not occur unless chromosomes are present. That same year, Walter Sutton (left) observed the separation of chromosomes into daughter cells during meiosis. Together, these observations led to the development of the Chromosomal Theory of Inheritance, which identified chromosomes as the genetic material responsible for Mendelian inheritance. The Chromosomal Theory of Inheritance was consistent with Mendel’s laws and was supported by the following observations: During meiosis, homologous chromosome pairs migrate as discrete structures that are independent of other chromosome pairs.