An Endogenous Foamy-Like Viral Element in the Coelacanth Genome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of a Full-Length Infectious Clone of Bovine Foamy Virus 3026
VIROLOGICA SINICA 2014, 29 (2): 94-102 DOI 10.1007/s12250-014-3382-5 RESEARCH ARTICLE Characterization of a full-length infectious clone of bovine foamy virus 3026 * Tiejun Bing1, Hong Yu1, 2, Yue Li1, Lei Sun3, Juan Tan1, Yunqi Geng1, Wentao Qiao1 1. Key Laboratory of Molecular Microbiology and Biotechnology (Ministry of Education) and Key Laboratory of Microbial Functional Genomics (Tianjin), College of Life Sciences, Nankai University, Tianjin 300071, China; 2. Department of Molecular and Cellular Pharmacology, the Vascular Biology Institute, University of Miami School of Medicine, Miami, FL 33136, USA; 3. National Laboratory of Biomacromolecules, Center for Biological Imaging, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China The biological features of most foamy viruses (FVs) are poorly understood, including bovine foamy virus (BFV). BFV strain 3026 (BFV3026) was isolated from the peripheral blood mononuclear cells of an infected cow in Zhangjiakou, China. A full-length genomic clone of BFV3026 was obtained from BFV3026-infected cells, and it exhibited more than 99% amino acid (AA) homology to another BFV strain isolated in the USA. Upon transfection into fetal canine thymus cells, the full-length BFV3026 clone produced viral structural and auxiliary proteins, typical cytopathic effects, and virus particles. These results demonstrate that the full-length BFV3026 clone is fully infectious and can be used in further BFV3026 research. KEYWORDS bovine foamy virus; infectious clone; syncytium; electron microscopy pol, and env structural genes and additional open read- INTRODUCTION ing frames (ORFs) that are under the control of the 5′- long terminal repeat (LTR) and an internal promoter (IP) Foamy viruses (FVs) are members of Retroviridae. -

Eukaryote Cell Biology - Michelle Gehringer
FUNDAMENTALS OF BIOCHEMISTRY, CELL BIOLOGY AND BIOPHYSICS – Vol. II - Eukaryote Cell Biology - Michelle Gehringer EUKARYOTE CELL BIOLOGY Michelle Gehringer Department of Biochemistry and Microbiology, University of Port Elizabeth, South Africa Keywords: cell theory, cell diversity, eukaryote cell structure, nucleus, chromatin, DNA, organelles, mitochondria, chloroplasts, transcription, RNA, translation, ribosomes, cell cycle, interphase, mitosis, meiosis, signal transduction, growth regulation, cancer, oncogenesis. Contents 1. Introduction 1.1. The first cell 2. Origin of Eukaryotes 3. Cellular differentiation in multicellular organisms 3.1. Plants 3.2. Animals 4. Eukaryotic cell structure 5. Organization of eukaryotic cells 5.1. Plasma membrane 5.2. Extracellular matrices 5.3. Protein synthesis and transport 5.4. Cytoskeleton and movement 5.5. Nucleus 5.5.1 Genomes 5.5.2 Gene expression 5.5.3 Maintaining the genome 5.6. Organelles 6. The cell cycle 6.1. Mitosis 6.2. Meiosis 7. Regulation of cell growth 7.1. Signal transduction 7.2. Programmed cell death 7.3. CancerUNESCO – EOLSS 8. Experimental Models 8.1. Yeast SAMPLE CHAPTERS 8.2. Arabidopsis 8.3. Drosophila 8.4. The mouse 8.5. Cell culture 8.6. Separation of cellular contents 8.7. Tracing biochemical pathways 9. Future Investigations Glossary Bibliography ©Encyclopedia of Life Support Systems (EOLSS) FUNDAMENTALS OF BIOCHEMISTRY, CELL BIOLOGY AND BIOPHYSICS – Vol. II - Eukaryote Cell Biology - Michelle Gehringer Biographical Sketch Summary Cells form the basic unit of life on our planet. They are well organized systems which perform all the essential tasks of eating, respiring, replicating and excreting waste products. The first cells, which are thought to have evolved about 3.8 billion years ago, much resembled present day prokaryotes. -

Feline Foamy Virus-Based Vectors: Advantages of an Authentic Animal Model
Viruses 2013, 5, 1702-1718; doi:10.3390/v5071702 OPEN ACCESS viruses ISSN 1999-4915 www.mdpi.com/journal/viruses Review Feline Foamy Virus-Based Vectors: Advantages of an Authentic Animal Model †,‡ † Weibin Liu , Janet Lei , Yang Liu, Dragana Slavkovic Lukic, Ann-Mareen Räthe, # Qiuying Bao, Timo Kehl, Anne Bleiholder, Torsten Hechler and Martin Löchelt * Department of Genome Modifications, Research Program Infection and Cancer, German Cancer Research Center, Im Neuenheimer Feld 242, 69120 Heidelberg, Germany; E-Mails: [email protected] (W.L.); [email protected] (J.L.); [email protected] (Y.L.); [email protected] (D.S.L.); [email protected] (A.-M.R.); [email protected] (Q.B.); [email protected] (T.K.); [email protected] (T.H.) † These authors contributed equally to this work. ‡ Current address: Department of Biochemistry and Molecular Biology, Mayo Clinic College of Medicine, 200 First Street SW, Rochester, MN 55905, USA. # Current address: Department of Biochemistry, Heidelberg Pharma GmbH, Schriesheimer Str. 101, 68526 Ladenburg, Germany. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +49-6221-424933; Fax: +49-6221-424932. Received: 1 March 2013; in revised form: 13 June 2013 / Accepted: 25 June 2013 / Published: 12 July 2013 Abstract: New-generation retroviral vectors have potential applications in vaccination and gene therapy. Foamy viruses are particularly interesting as vectors, because they are not associated to any disease. Vector research is mainly based on primate foamy viruses (PFV), but cats are an alternative animal model, due to their smaller size and the existence of a cognate feline foamy virus (FFV). -

Characterization of Biofilm Extracts from Two Marine Bacteria
applied sciences Article Characterization of Biofilm Extracts from Two Marine Bacteria Delphine Passerini 1, Florian Fécamp 1, Laetitia Marchand 1, Laetitia Kolypczuk 1 , Sandrine Bonnetot 1, Corinne Sinquin 1 ,Véronique Verrez-Bagnis 1 , Dominique Hervio-Heath 2 , Sylvia Colliec-Jouault 1 and Christine Delbarre-Ladrat 1,* 1 Ifremer, Atlantique Center, Microbial Ecosystems and Marine Molecules for the Biotechnologies, 44311 Rue de l’Ile d’Yeu, Nantes CEDEX 3, France; [email protected] (D.P.); fl[email protected] (F.F.); [email protected] (L.M.); [email protected] (L.K.); [email protected] (S.B.); [email protected] (C.S.); [email protected] (V.V.-B.); [email protected] (S.C.-J.) 2 Ifremer, Bretagne Center, Health, Environment and Microbiology, 1625 Route de Sainte-Anne, 29280 Plouzané, France; [email protected] * Correspondence: [email protected] Received: 17 September 2019; Accepted: 13 November 2019; Published: 19 November 2019 Featured Application: By analyzing extracts of biofilm formed by two marine bacteria and comparing them with planktonic extracts, we have shown that biofilm may induce the biosynthesis of potentially bioactive compounds and may open up new possibilities for compound discovery. Abstract: In the marine environment, biofilm formation is an important lifestyle for microorganisms. A biofilm is comprised of cells embedded in an extracellular matrix that holds them close together and keeps the biofilm attached to the colonized surface. This predominant lifestyle and its main regulation pathway, namely quorum-sensing (QS), have been shown to induce specific bioactive metabolites. In this study, we investigated the biofilm formation by two marine bacteria belonging to the Vibrio species to discover potentially innovative bioactive compounds. -

The Origin of the Eukaryotic Cell Based on Conservation of Existing
The Origin of the Eukaryotic Albert D. G. de Roos The Beagle Armada Cell Based on Conservation Bioinformatics Division of Existing Interfaces Einsteinstraat 67 3316GG Dordrecht, The Netherlands [email protected] Abstract Current theories about the origin of the eukaryotic Keywords cell all assume that during evolution a prokaryotic cell acquired a Evolution, nucleus, eukaryotes, self-assembly, cellular membranes nucleus. Here, it is shown that a scenario in which the nucleus acquired a plasma membrane is inherently less complex because existing interfaces remain intact during evolution. Using this scenario, the evolution to the first eukaryotic cell can be modeled in three steps, based on the self-assembly of cellular membranes by lipid-protein interactions. First, the inclusion of chromosomes in a nuclear membrane is mediated by interactions between laminar proteins and lipid vesicles. Second, the formation of a primitive endoplasmic reticulum, or exomembrane, is induced by the expression of intrinsic membrane proteins. Third, a plasma membrane is formed by fusion of exomembrane vesicles on the cytoskeletal protein scaffold. All three self-assembly processes occur both in vivo and in vitro. This new model provides a gradual Darwinistic evolutionary model of the origins of the eukaryotic cell and suggests an inherent ability of an ancestral, primitive genome to induce its own inclusion in a membrane. 1 Introduction The origin of eukaryotes is one of the major challenges in evolutionary cell biology. No inter- mediates between prokaryotes and eukaryotes have been found, and the steps leading to eukaryotic endomembranes and endoskeleton are poorly understood. There are basically two competing classes of hypotheses: the endosymbiotic and the autogenic. -

Study of Chikungunya Virus Entry and Host Response to Infection Marie Cresson
Study of chikungunya virus entry and host response to infection Marie Cresson To cite this version: Marie Cresson. Study of chikungunya virus entry and host response to infection. Virology. Uni- versité de Lyon; Institut Pasteur of Shanghai. Chinese Academy of Sciences, 2019. English. NNT : 2019LYSE1050. tel-03270900 HAL Id: tel-03270900 https://tel.archives-ouvertes.fr/tel-03270900 Submitted on 25 Jun 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. N°d’ordre NNT : 2019LYSE1050 THESE de DOCTORAT DE L’UNIVERSITE DE LYON opérée au sein de l’Université Claude Bernard Lyon 1 Ecole Doctorale N° 341 – E2M2 Evolution, Ecosystèmes, Microbiologie, Modélisation Spécialité de doctorat : Biologie Discipline : Virologie Soutenue publiquement le 15/04/2019, par : Marie Cresson Study of chikungunya virus entry and host response to infection Devant le jury composé de : Choumet Valérie - Chargée de recherche - Institut Pasteur Paris Rapporteure Meng Guangxun - Professeur - Institut Pasteur Shanghai Rapporteur Lozach Pierre-Yves - Chargé de recherche - CHU d'Heidelberg Rapporteur Kretz Carole - Professeure - Université Claude Bernard Lyon 1 Examinatrice Roques Pierre - Directeur de recherche - CEA Fontenay-aux-Roses Examinateur Maisse-Paradisi Carine - Chargée de recherche - INRA Directrice de thèse Lavillette Dimitri - Professeur - Institut Pasteur Shanghai Co-directeur de thèse 2 UNIVERSITE CLAUDE BERNARD - LYON 1 Président de l’Université M. -

Molecular Analysis of the Complete Genome of a Simian Foamy Virus Infecting Hylobates Pileatus (Pileated Gibbon) Reveals Ancient Co-Evolution with Lesser Apes
viruses Article Molecular Analysis of the Complete Genome of a Simian Foamy Virus Infecting Hylobates pileatus (pileated gibbon) Reveals Ancient Co-Evolution with Lesser Apes Anupama Shankar 1, Samuel D. Sibley 2, Tony L. Goldberg 2 and William M. Switzer 1,* 1 Laboratory Branch, Division of HIV/AIDS Prevention, Center for Disease Control and Prevention, Atlanta, GA 30329, USA 2 Department of Pathobiological Sciences, School of Veterinary Medicine, University of Wisconsin-Madison, Madison, WI 53706, USA * Correspondence: [email protected]; Tel.: +1-404-639-2019 Received: 1 April 2019; Accepted: 30 June 2019; Published: 3 July 2019 Abstract: Foamy viruses (FVs) are complex retroviruses present in many mammals, including nonhuman primates, where they are called simian foamy viruses (SFVs). SFVs can zoonotically infect humans, but very few complete SFV genomes are available, hampering the design of diagnostic assays. Gibbons are lesser apes widespread across Southeast Asia that can be infected with SFV, but only two partial SFV sequences are currently available. We used a metagenomics approach with next-generation sequencing of nucleic acid extracted from the cell culture of a blood specimen from a lesser ape, the pileated gibbon (Hylobates pileatus), to obtain the complete SFVhpi_SAM106 genome. We used Bayesian analysis to co-infer phylogenetic relationships and divergence dates. SFVhpi_SAM106 is ancestral to other ape SFVs with a divergence date of ~20.6 million years ago, reflecting ancient co-evolution of the host and SFVhpi_SAM106. Analysis of the complete SFVhpi_SAM106 genome shows that it has the same genetic architecture as other SFVs but has the longest recorded genome (13,885-nt) due to a longer long terminal repeat region (2,071 bp). -

Sensitivity of Golgi Matrix Proteins to an ER Exit Block
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central JCBArticle Evidence that the entire Golgi apparatus cycles in interphase HeLa cells: sensitivity of Golgi matrix proteins to an ER exit block Suzanne Miles,1 Heather McManus,1 Kimberly E. Forsten,2 and Brian Storrie1 1Department of Biochemistry and 2Department of Chemical Engineering, Virginia Tech, Blacksburg, VA 24061 e tested whether the entire Golgi apparatus is concentration. Redistribution of GalNAcT2 was more a dynamic structure in interphase mammalian sensitive to low Sar1pdn concentrations than giantin or Wcells by assessing the response of 12 different GM130. Redistribution was most rapid for p27, COPI, and Golgi region proteins to an endoplasmic reticulum (ER) exit p115. Giantin, GM130, and GalNAcT2 relocated with block. The proteins chosen spanned the Golgi apparatus approximately equal kinetics. Distinct ER accumulation and included both Golgi glycosyltransferases and putative could be demonstrated for all integral membrane proteins. matrix proteins. Protein exit from ER was blocked either by ER-accumulated Golgi region proteins were functional. microinjection of a GTP-restricted Sar1p mutant protein in Photobleaching experiments indicated that Golgi-to-ER the presence of a protein synthesis inhibitor, or by plasmid- protein cycling occurred in the absence of any ER exit encoded expression of the same dominant negative Sar1p. block. We conclude that the entire Golgi apparatus is a All Golgi region proteins examined lost juxtanuclear Golgi dynamic structure and suggest that most, if not all, Golgi apparatus–like distribution as scored by conventional and region–integral membrane proteins cycle through ER in confocal fluorescence microscopy in response to an ER interphase cells. -

Center of Biomedical Research Excellence in Matrix Biology: Building Research Infrastructure, Supporting Young Researchers, and Fostering Collaboration
International Journal of Molecular Sciences Editorial Center of Biomedical Research Excellence in Matrix Biology: Building Research Infrastructure, Supporting Young Researchers, and Fostering Collaboration Julia Thom Oxford 1,2,3,* , Ken A. Cornell 1,3,4, Jared J. Romero 1,5 , Diane B. Smith 1,3 , Tracy L. Yarnell 1,3, Rhiannon M. Wood 1,3 , Cheryl L. Jorcyk 1,2, Trevor J. Lujan 1,6, Allan R. Albig 1,2 , Kristen A. Mitchell 1,2, Owen M. McDougal 1,4 , Daniel Fologea 1,7, David Estrada 1,8, Juliette K. Tinker 1,2, Rajesh Nagarajan 1,4, Don L. Warner 1,4, Troy T. Rohn 1,2, Jim Browning 1,9 , Richard S. Beard Jr. 1,3, Lisa R. Warner 1,4 , Brad E. Morrison 1,2, Clare K. Fitzpatrick 1,6, Gunes Uzer 1,6, Laura Bond 1,3, Stephanie M. Frahs 1,3, Cynthia Keller-Peck 1,3, Xinzhu Pu 1,3, Luke G. Woodbury 1,3 and Matthew W. Turner 1,3 1 Center of Biomedical Research Excellence in Matrix Biology, Boise State University, Boise, ID 83725, USA 2 Department of Biological Sciences, Boise State University, Boise, ID 83725, USA 3 Biomolecular Research Center, Boise State University, Boise, ID 83725, USA 4 Department of Chemistry and Biochemistry, Boise State University, Boise, ID 83725, USA 5 Division of Research, Boise State University, Boise, ID 83725, USA 6 Department of Mechanical and Biomedical Engineering, Boise State University, Boise, ID 83725, USA 7 Department of Physics, Boise State University, Boise, ID 83725, USA 8 Micron School of Materials Science and Engineering, Boise State University, Boise, ID 83725, USA 9 Department of Electrical and Computer Engineering, Boise State University, Boise, ID 83725, USA * Correspondence: [email protected]; Tel.: +01-208-426-2395 Received: 9 March 2020; Accepted: 17 March 2020; Published: 20 March 2020 Abstract: The Center of Biomedical Research Excellence in Matrix Biology strives to improve our understanding of extracellular matrix at molecular, cellular, tissue, and organismal levels to generate new knowledge about pathophysiology, normal development, and regenerative medicine. -
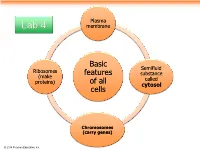
Basic Features of All Cells
Plasma Lab 4 membrane Basic Semifluid Ribosomes features substance (make called proteins) of all cytosol cells Chromosomes (carry genes) © 2014 Pearson Education, Inc. Prokaryotic cells are characterized by having . No nucleus . DNA in an unbound region called the nucleoid . No membrane-bound organelles . Cytoplasm bound by the plasma membrane © 2014 Pearson Education, Inc. Eukaryotic cells are characterized by having • DNA in a nucleus that is bounded by a membranous nuclear envelope • Membrane-bound organelles • Cytoplasm in the region between the plasma membrane and nucleus Eukaryotic cells are generally much larger than prokaryotic cells © 2014 Pearson Education, Inc. Figure 6.8a ENDOPLASMIC RETICULUM (ER) Nuclear envelope Rough ER Smooth ER Nucleolus NUCLEUS Flagellum Chromatin Centrosome Plasma membrane CYTOSKELETON: Microfilaments Intermediate filaments Microtubules Ribosomes Microvilli Golgi apparatus Peroxisome Lysosome Mitochondrion © 2014 Pearson Education, Inc. Figure 6.8b Nuclear envelope NUCLEUS Nucleolus Rough ER Chromatin Smooth ER Ribosomes Golgi Central vacuole apparatus Microfilaments CYTOSKELETON Microtubules Mitochondrion Peroxisome Plasma Chloroplast membrane Cell wall Plasmodesmata Wall of adjacent cell © 2014 Pearson Education, Inc. © 2014 Pearson Education, Inc. © 2014 Pearson Education, Inc. © 2014 Pearson Education, Inc. Table 6.1 © 2014 Pearson Education, Inc. Cell Walls of Plants The cell wall is an extracellular structure that distinguishes plant cells from animal cells Plant cell walls Prokaryotes, are made -

Analysis of the Role of the Bel and Bet Open Reading Frames of Human Foamy Virus by Using a New Quantitative Assay SHUYUARN F
JOURNAL OF VIROLOGY, Nov. 1993, p. 6618-6624 Vol. 67, No. 11 0022-538X/93/1 16618-07$02.00/0 Copyright C 1993, American Society for Microbiology Analysis of the Role of the bel and bet Open Reading Frames of Human Foamy Virus by Using a New Quantitative Assay SHUYUARN F. YU AND MAXINE L. LINIAL* Division of Basic Sciences, Fred Hutchinson Cancer Research Center, 1124 Columbia Street, Seattle, Washington 98104-2092 Received 6 July 1993/Accepted 5 August 1993 We have constructed a BHK-21-derived indicator cell line containing a single integrated copy of the f-galactosidase (P-Gal) gene under control of the human foamy virus (HFV) long terminal repeat promoter (from -533 to +20). These foamy virus-activated ,8-Gal expression (FAB) cells can be used in a quantitative assay to measure the infectious titer of HFV. Our results show that the FAB assay is 50 times more sensitive than determination of the virus titer by the end-point dilution method. Using the FAB assay, we have found that HFV can productively replicate in several erythroblastoid cell lines as well as in the Jurkat T-cell line. We have also examined the roles of bel2, bet, and beI3 in viral replication by constructing proviral HFV clones in which the reading frame of Bel2, Bet, or Bel3 is disrupted by placement of translation stop codons. Analysis of these mutants reveals that while the beI3 gene is not required for viral replication in vitro, mutations in the beI2 or bet gene decrease cell-free viral transmission approximately 10-fold. -

Characterization of Giardia Cell Nucleus: Its Implication on the Nature and Origin of the Primitive Cell Nucleus
Cell Research(1995),5,115—124 Characterization of Giardia cell nucleus: Its implication on the nature and origin of the primitive cell nucleus LI JINGYAN Department of Evolutionary Cell Biology, Laboratory of Cellular and Molecular Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China. Key words: Origin of cell nucleus, nucleus of Giardia, primitive cell nu- cleus, archezoa. The central problem of the origin and early evolution of the eukaryotic cell is the origin of the cell nucleus, because the cell nucleus, with its double-layered nuclear envelope, is the most prominent and important morphological mark distinguishing eukaryotic cells from prokaryotes. Eukaryotic cells may have no chloroplast, mito- chondria or flagellum, but they must possess a cell nucleus. The cell nucleus itself is a very complicated structure. If we try to make clear the origin and early evolution of the cell nucleus, we ought to investigate the origin and evolution of each morphological component of a cell nucleus. My monograph on‘The genesis of eukaryotic cells in the evolutionary history of life’(1979, Science Press, Beijing, in Chinese)[1], mainly attempted to make some effort in this regard. However, to date, the hypothetical model of a fully depicted nucleus of a primitive cell has not been constructed. It seems likely that our recent studies of the nucleus of Giardia (diplomonad) have provided a realistic basis to construct such a model. I. The characteristics of the nucleus of Giardia Kingdom Archezoa (Cavalier-Smith, 1989), contains those parasitic and free- living protists which originally possess no mitochondria and no typical Golgi ap- paratus, but they have a 70s type of ribosomes containing 16s and 23s rRNAs, just as prokaryotes have.