Download (6MB)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluated Gas Phase Basicities and Proton Affinities of Molecules; Heats of Formation of Protonated Molecules
Evaluated Gas Phase Basicities and Proton Affinities of Molecules; Heats of Formation of Protonated Molecules Cite as: Journal of Physical and Chemical Reference Data 13, 695 (1984); https://doi.org/10.1063/1.555719 Published Online: 15 October 2009 Sharon G. Lias, Joel F. Liebman, and Rhoda D. Levin ARTICLES YOU MAY BE INTERESTED IN Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update Journal of Physical and Chemical Reference Data 27, 413 (1998); https:// doi.org/10.1063/1.556018 Density-functional thermochemistry. III. The role of exact exchange The Journal of Chemical Physics 98, 5648 (1993); https://doi.org/10.1063/1.464913 A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu The Journal of Chemical Physics 132, 154104 (2010); https://doi.org/10.1063/1.3382344 Journal of Physical and Chemical Reference Data 13, 695 (1984); https://doi.org/10.1063/1.555719 13, 695 © 1984 American Institute of Physics for the National Institute of Standards and Technology. Evaluated Gas Phase Basicities and Proton Affinities of Molecules; Heats of Formation of Protonated Molecules Sharon G. Lias Center for Chemical Physics, National Bureau of Standards, Gaithersburg, MD 20899 Joel F. Liebman Department of Chemistry, University of Maryland Baltimore County, Catonsville, MD 21228 and Rhoda D. Levin Center for Chemical Physics. National Bureau of Standards, Gaithersburg, MD 20899 The available data on gas phase basicities and proton affinities of molecules are compiled and evaluated. Tables giving the molecules ordered (1) according to proton affinity and (2) according to empirical formula, sorted alphabetically are provided. -

Intramolecular Ene Reactions of Functionalised Nitroso Compounds
INTRAMOLECULAR ENE REACTIONS OF FUNCTIONALISED NITROSO COMPOUNDS A Thesis Presented by Sandra Luengo Arratta In Partial Fulfilment of the Requirements for the Award of the Degree of DOCTOR OF PHILOSOPHY OF UNIVERSITY COLLEGE LONDON Christopher Ingold Laboratories Department of Chemistry University College London 20 Gordon Street London WC1H 0AJ July 2010 DECLARATION I Sandra Luengo Arratta, confirm that the work presented in this thesis is my own. Where work has been derived from other sources, I confirm that this has been indicated in the thesis. ABSTRACT This thesis concerns the generation of geminally functionalised nitroso compounds and their subsequent use in intramolecular ene reactions of types I and II, in order to generate hydroxylamine derivatives which can evolve to the corresponding nitrones. The product nitrones can then be trapped in the inter- or intramolecular mode by a variety of reactions, including 1,3-dipolar cycloadditions, thereby leading to diversity oriented synthesis. The first section comprises the chemistry of the nitroso group with a brief discussion of the current methods for their generation together with the scope and limitations of these methods for carrying out nitroso ene reactions, with different examples of its potential as a powerful synthetic method to generate target drugs. The second chapter describes the results of the research programme and opens with the development of methods for the generation of functionalised nitroso compounds from different precursors including oximes and nitro compounds, using a range of reactants and conditions. The application of these methods in intramolecular nitroso ene reactions is then discussed. Chapter three presents the conclusions which have been drawn from the work presented in chapter two, and provides suggestions for possible directions of this research in the future. -
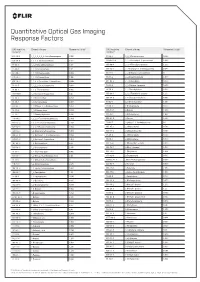
Quantitative Optical Gas Imaging Response Factors
Quantitative Optical Gas Imaging Response Factors CAS registry Chemical name Response factor† CAS registry Chemical name Response factor† number* number* 431-89-0 1_1_1_2_3_3_3-Heptafluoropropane 0.103 75-89-8 2_2_2-Trifluoroethanol 0.326 630-20-6 1_1_1_2-Tetrachloroethane 0.132 25256-77-4 2_2_4-Trimethyl-1_3-pentanediol 1.086 75-68-3 1_1_1-Chlorodifluoroethane 0.117 107-40-4 2_2_4-Trimethyl-2-pentene 1.262 71-55-6 1_1_1-Trichloroethane 0.121 306-83-2 2_2-Dichloro-1_1_1-trifluoroethane 0.047 421-50-1 1_1_1-Trifluoroacetone 0.086 76-11-9 2_2-Difluorotetrachloroethane 0 420-46-2 1_1_1-Trifluoroethane 0.106 75-83-2 2_2-Dimethyl_butane 1.944 354-14-3 1_1_2_2-Tetrachloro-1-fluoroethane 0.046 431-03-8 2_3-Butanedione 0.274 79-34-5 1_1_2_2-Tetrachloroethane 0.069 78-88-6 2_3-Dichloro-1-propene 0.279 79-00-5 1_1_2-Trichloroethane 0.102 79-29-8 2_3-Dimethylbutane 1.014 1717-00-6 1_1-Dichloro-1-fluoroethane 0.12 107-39-1 2_4_4-Trimethyl-1-pentene 1.212 75-34-3 1_1-Dichloroethane 0.272 584-84-9 2_4-Diisocyanatetoluene 0.895 75-35-4 1_1-Dichloroethene 0.017 87-62-7 2_6-Dimethylaniline 1.308 471-43-2 1_1-Difluoro-2_2-dichloroethane 0.352 75-26-3 2-Bromopropane 0.733 73-37-6 1_1-Difluoroethane 0.535 107-01-7 2-Butene 1.204 57-14-7 1_1-Dimethylhydrazine 0.897 111-76-2 2-Butoxyethanol 1.962 119-64-2 1_2_3_4-Tetrahydronaphthalene 2.439 554-61-0 2-Carene 1.286 488-23-3 1_2_3_4-Tetramethylbenzene 1.484 75-88-7 2-Chloro-1_1_1-trifluoroethane 0.1 527-53-7 1_2_3_5-Tetramethylbenzene 1.402 107-07-3 2-Chloroethanol 0.593 124-73-2 1_2-Dibomotetrafluoroethane -

PROLIFERATION: THREAT and RESPONSE January 2001
PROLIFERATION: THREAT AND RESPONSE January 2001 Office of the Secretary of Defense NT OF E D M E T F R E A N P S E E D U N A I C T I E R D E S T A M AT E S O F Proliferation: Threat and Response January 2001 This document, along with other DoD materials related to nonproliferation and counterproliferation, may be found at http://www.defenselink.mil Message of the Secretary of Defense At the dawn of the 21st Century, the United States now faces what could be called a Superpower Paradox. Our unrivaled supremacy in the conventional military arena is prompting adversaries to seek unconventional, asymmetric means to strike what they perceive as our Achilles heel. At least 25 countries now possess — or are in the process of acquiring and developing — capabilities to inflict mass casualties and destruction: nuclear, biologi- cal and chemical (NBC) weapons or the means to deliver them. For example: I North Korea is building and selling long-range missiles, has chemical and biologi- cal warfare capabilities, and may have diverted fissile material for nuclear weap- onry. I Iran, with foreign assistance, is buying and developing longer-range missiles, already has chemical weapons, and is seeking nuclear and biological capabilities. I Iraq — which prior to the 1991 Gulf War had developed chemical and biological weapons and associated delivery means, and was close to a nuclear capability — may have reconstituted these efforts since the departure of UN inspectors from Iraq in late 1998. I Libya has chemical capabilities and is trying to buy long-range missiles. -

Supplementary Information Cocrystal Design by Network-Based Link Prediction
Electronic Supplementary Material (ESI) for CrystEngComm. This journal is © The Royal Society of Chemistry 2019 Supplementary Information Cocrystal design by network-based link prediction Jan-Joris Devogelaer, Sander J.T. Brugman, Hugo Meekes, Paul Tinnemans, Elias Vlieg, René de Gelder Table of Contents S1 Materials and syntheses page 1 S2 Single-crystal X-ray structure determinations, ORTEP plots and H-Bonding page 6 a) CCDC 1940959 = p1812d: 4,4’-bipyridine + resorcinol b) CCDC 1940950 = p1823c: 1,2-di(4-pyridyl)ethylene + 4-aminobenzoic acid c) CCDC 1940956 = p1823a: 1,2-di(4-pyridyl)ethylene + 4-aminobenzoic acid dihydrate d) CCDC 1940954 = p1907a: 1,2-di(4-pyridyl)ethylene + sebacic acid e) CCDC 1940949 = p1818a: 4,4’-bipyridine + suberic acid f) CCDC 1940953 = p1922b: 1,2-di(4-pyridyl)ethylene + oxalic acid g) CCDC 1940955 = p1914b: 1,2-di(4-pyridyl)ethylene + oxalic acid dihydrate i) CCDC 1940951 = p1908a: 1,2-bis(4-pyridyl)ethane + salicylic acid j) CCDC 1940958 = p1926b: 1,2-di(4-pyridyl)ethylene + 4-nitrobenzoic acid k) CCDC 1940952 = p1910a: 1,2-di(4-pyridyl)ethylene + phthalic acid l) CCDC 1940957 = p1913a: 1,2-di(4-pyridyl)ethylene + malonic acid S3 Top 100 predictions of the scoring method page 17 S4 Predefined lists of common solvents and gases page 21 S1 - Materials and syntheses We first present the materials in Table S1, after which we discuss the protocol used to obtain crystals suitable for single-crystal X-ray diffraction. Besides the two-component structures shown in Table 2, an additional cocrystal dihydrate (c) and salt dihydrate (g) were found. -

Enthalpies of Vaporization of Organic and Organometallic Compounds, 1880–2002
Enthalpies of Vaporization of Organic and Organometallic Compounds, 1880–2002 James S. Chickosa… Department of Chemistry, University of Missouri-St. Louis, St. Louis, Missouri 63121 William E. Acree, Jr.b… Department of Chemistry, University of North Texas, Denton, Texas 76203 ͑Received 17 June 2002; accepted 17 October 2002; published 21 April 2003͒ A compendium of vaporization enthalpies published within the period 1910–2002 is reported. A brief review of temperature adjustments of vaporization enthalpies from temperature of measurement to the standard reference temperature, 298.15 K, is included as are recently suggested reference materials. Vaporization enthalpies are included for organic, organo-metallic, and a few inorganic compounds. This compendium is the third in a series focusing on phase change enthalpies. Previous compendia focused on fusion and sublimation enthalpies. Sufficient data are presently available for many compounds that thermodynamic cycles can be constructed to evaluate the reliability of the measure- ments. A protocol for doing so is described. © 2003 American Institute of Physics. ͓DOI: 10.1063/1.1529214͔ Key words: compendium; enthalpies of condensation; evaporation; organic compounds; vaporization enthalpy. Contents inorganic compounds, 1880–2002. ............. 820 1. Introduction................................ 519 8. References to Tables 6 and 7.................. 853 2. Reference Materials for Vaporization Enthalpy Measurements.............................. 520 List of Figures 3. Heat Capacity Adjustments. ................. 520 1. A thermodynamic cycle for adjusting vaporization ϭ 4. Group Additivity Values for C (298.15 K) enthalpies to T 298.15 K.................... 521 pl 2. A hypothetical molecule illustrating the different Estimations................................ 521 hydrocarbon groups in estimating C ........... 523 5. A Thermochemical Cycle: Sublimation, p Vaporization, and Fusion Enthalpies........... -

CBW Conventions Bulletin 51
THE CBW CONVENTIONS BULLETIN News, Background and Comment on Chemical and Biological Weapons Issues ISSUE NO. 51 MARCH 2001 Quarterly Journal of the Harvard Sussex Program on CBW Armament and Arms Limitation WASHINGTON AND THE BWC PROTOCOL NEGOTIATION In the aftermath of the February session of the BWC Ad decision, the deadline for completion will not be met, as Hoc Group, on which Graham Pearson reports in this issue, there will not be time enough to address the outstanding it is far from clear that the BWC Protocol will be completed substantive and procedural issues between then and before the commencement of the Fifth BWC Review November. Conference in November 2001. If, on the other hand, Ambassador Tóth goes forward What is clear, however, is that decisions taken between with the composite text, the Protocol’s prospects in 2001 now and the April Preparatory Committee for the Review may well depend upon decisions taken by key Non-Aligned Conference will be critical in determining whether that Movement (NAM) and other countries. deadline — agreed by states parties at their Fourth Review Nowhere is this more true than on the issue of export Conference, in 1996 — is met. controls, which has pitted Iran and some other NAM The first and potentially most important decision is to be countries against the USA and others in the West. Iran has made in the United States, where the new administration of heretofore argued that existing multilateral export controls President George W Bush has launched what is reported to on biological materials are discriminatory and that, if it is to be a broad review of US policy toward the BWC Protocol. -

Phosgene Oxime
Phosgene oxime From Wikipedia, the free encyclopedia Jump to navigation Jump to search Phosgene oxime Names IUPAC name N-(dichloromethylidene)hydroxylamine Other names dichloroformaldoxime, dichloroformoxime, hydroxycarbonimidic dichloride, CX Identifiers Y CAS Number • 1794-86-1 • Interactive image 3D model (JSmol) ChemSpider • 59024 Y • 65582 PubChem CID UNII • G45S3149SQ Y • DTXSID9075292 CompTox Dashboard (EPA) show InChI • InChI=1S/CHCl2NO/c2-1(3)4-5/h5H Y Key: JIRJHEXNDQBKRZ-UHFFFAOYSA-N Y • InChI=1/CHCl2NO/c2-1(3)4-5/h5H Key: JIRJHEXNDQBKRZ-UHFFFAOYAP show SMILES • Cl/C(Cl)=N\O Properties CHCl NO Chemical formula 2 Molar mass 113.93 g·mol−1 Appearance colorless crystalline solid or yellowish-brown liquid[1] Melting point 35 to 40 °C (95 to 104 °F; 308 to 313 K)[1] Boiling point 128 °C (262 °F; 401 K)[1] 70%[1] Solubility in water Hazards Main hazards highly toxic Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). N verify (what is Y N ?) Infobox references Chemical compound Phosgene oxime, or CX, is an organic compound with the formula Cl2CNOH. It is a potent chemical weapon, specifically a nettle agent. The compound itself is a colorless solid, but impure samples are often yellowish liquids. It has a strong, disagreeable odor and a violently irritating vapor. Contents • 1 Preparation and reactions • 2 Safety • 2.1 Decontamination, treatment, and handling properties • 3 References • 4 External links Preparation and reactions[edit] Phosgene oxime can be prepared by reduction of chloropicrin using a combination of tin metal and hydrochloric acid as the source of the active hydrogen reducing acent: Cl3CNO2 + 4 [H] → Cl2C=N−OH + HCl + H2O The observation of a transient violet color in the reaction suggests intermediate formation of trichloronitrosomethane (Cl3CNO). -

Ep 0128006 B1
Europaisches Paten tamt J European Patent Office C11) Publication number: 0 128 006 B1 Office europeen des brevets EUROPEAN PATENT SPECIFICATION © Date of publication of patent specification: 18.09.91 © Int. Cl.5: C07C 211/39, C07C 229/38, C07C 249/02, C07C 255/24, © Application number: 84303640.1 C07C 257/00, C07C 233/00, C07D 227/00, C07D 307/68, ©@r\n* Date of,«■ f,,,ng:filing: 30.05.84onnco, ^ ^ £ ^ ^^ C07D 251/40 A soil-disease-controlling agent. ® Priority: 31.05.83 JP 97545/83 03.06.83 JP 99959/83 (73) Proprietor: SUMITOMO CHEMICAL COMPANY, 17.08.83 JP 150853/83 LIMITED 18.08.83 JP 151117/83 Kitahama 4-chome 5-33 13.06.83 JP 106233/83 Chuo-ku Osaka 541 (JP) 09.06.83 JP 103949/83 @ Inventor: Tomioka, Hiroki © Date of publication of application: 2-14-7, Mefu 12.12.84 Bulletin 84/50 Takarazuka Hyogo-ken(JP) Inventor: Ooishi, Tadashi © Publication of the grant of the patent: 2-10-2-231, Sonehigashimachi 18.09.91 Bulletin 91/38 Toyonaka Osaka-fu(JP) Inventor: Takahashi, Junya ® Designated Contracting States: 4-2-303, Ryodocho CH DE FR GB IT LI NL Nishinomiya Hyogo-ken(JP) Inventor: Sasaki, Mitsuru References cited: 2-10-1-112, Sonehigashimachi CO DE-A- 1 956 285 GB-A- 806 798 Toyonaka Osaka-fu(JP) CO GB-A- 2 061 913 US-A- 2 512 293 Inventor: Hirata, Naonori o US-A- 3 385 846 US-A- 3 494 105 704-2, 7-510 Kanaokacho o US-A- 3 574 746 US-A- 3 679 699 Sakai Osaka-fu(JP) 00 US-A- 4 204 997 Note: Within nine months from the publication of the mention of the grant of the European patent, any person notice the Patent Office of the CL may give to European opposition to European patent granted. -

Overall View of Chemical and Biochemical Weapons
Toxins 2014, 6, 1761-1784; doi:10.3390/toxins6061761 OPEN ACCESS toxins ISSN 2072-6651 www.mdpi.com/journal/toxins Review Overall View of Chemical and Biochemical Weapons Vladimír Pitschmann 1,2 1 Faculty of Biomedical Engineering, Czech Technical University in Prague, nám. Sítná 3105, Kladno 27201, Czech Republic 2 Oritest spol. s r.o., Staropramenná 17, Prague 15000, Czech Republic; E-Mail: [email protected]; Tel.: +420-257-311-639 Received: 11 April 2014; in revised form: 15 May 2014 / Accepted: 26 May 2014 / Published: 4 June 2014 Abstract: This article describes a brief history of chemical warfare, which culminated in the signing of the Chemical Weapons Convention. It describes the current level of chemical weapons and the risk of using them. Furthermore, some traditional technology for the development of chemical weapons, such as increasing toxicity, methods of overcoming chemical protection, research on natural toxins or the introduction of binary technology, has been described. In accordance with many parameters, chemical weapons based on traditional technologies have achieved the limit of their development. There is, however, a big potential of their further development based on the most recent knowledge of modern scientific and technical disciplines, particularly at the boundary of chemistry and biology. The risk is even higher due to the fact that already, today, there is a general acceptance of the development of non-lethal chemical weapons at a technologically higher level. In the future, the chemical arsenal will be based on the accumulation of important information from the fields of chemical, biological and toxin weapons. Data banks obtained in this way will be hardly accessible and the risk of their materialization will persist. -

Some Design Considerations, Large Expulsion Bladders for Nitrogen Tetroxide and Hydrazine
NATIONAL AERONAUTICS AND SPACE ADMINISTRATION Technics/ Report No. 32-862 Some Design Considerations, Large Expulsion Bladders for Nitrogen Tetroxide and Hydrazine A. J. Bauman , i (THRUI t / 1 3(T I (NASA CR OR TMX OR AD'NUMBER) (CATEGORY1 I I GPO PRICE $ I CFSTl PRICE(S) $ Hard copy (HC) %2 I 0 I ', Microfiche (MF) 13% ,< ff 653 July 65 JET PROPULSION LABORATORY CALIFORNIA lNSTlTUTE OF TECHNOLOGY PASADENA,CALIFORNIA January 15, 1966 NATIONAL AERONAUTICS AND SPACE ADMINISTRATION Technical Report No. 32-862 Some Design Considerations, Large Expulsion Bladders for Nitrogen Tetroxide and Hydrazine A. J. Bauman D. F. Dipprey, Manager liquid Propulsion Section JET PROPULSION LABORATORY CALIFORNIA INSTITUTE OF TECHNOLOGY PASADENA,CALI FOR N IA January 15, 1966 Copyright 0 1966 Jet Propulsion Laboratory California Institute of Technology Prepared Under Contract No. NAS 7-100 National Aeronautics & Space Administration JPL TECHNICAL REPORT NO. 32-862 CONTENTS 1 . Introduction .................... 1 II. The ALPS Bladder Problem ............... 1 111. N.O. Compatibility ................. 2 IV. Properties of Fuel and Oxidizer ............. 3 V . The Stanford-Vango Experiment ............ 4 VI. Critical Hypergolicity Limits .............. 5 VI1. leafing Pigments and Filled Membranes ......... 6 VIII . Removal Systems .................. 7 IX. Bag Folding .................... 8 X . An Advantageous Design Modification ......... 10 XI. Conclusions .................... 10 Appendixes A . The Chemical Stability of High Polymers Toward Oxidizing Agents .............11 B. Permeation Through Membranes ..........20 C . Teflon ..................... 21 References .......................22 Bibliography ......................28 A.1 . Chemical structures and trade names of fluoropolymers ..... 13 A.2 . Typical physical properties of fluoropolymers .........13 A.3 . Resistance of typical fluoroelastomers to chemicals and solvents ...14 A.4 . Miscellaneous fluoropolymers ............. -

Table 1A. Standard (Based on ISO and ASTM) Abbreviations of Plastics
491 TABLE 1A Table 1a. Standard (based on ISO and ASTM) abbreviations ofplastics Abbrevi Name Abbrevi Name ation ation ABS Acrylonitrile butadiene styrene. PMP Poly-4-methylpentene-l. NB/A Acrylonitrile/butadiene/acrylate. PMS Poly-a-methylstyrene. NCPE/S Acrylonitrile/chlorinated polyethylene/styrene. POM Polyoxymethylene or, poly acetal NEPDM/S Acrylonitrile/ethylene-propylene-diene/styrene. or, polyformaldehyde. NMMA Acrylonitrile/methyl methacrylate. PP Polypropylene. ASA Acrylonitrile/styrene/acrylate. PPE Polyphenylene ether. CA Cellulose acetate. PPOX Polypropylene oxide. CAB Cellulose acetate butyrate. PPS Polyphenylene sulphide. CAP Cellulose acetate propionate. PPSU Polyphenylene sulphone. CF Cresol-formaldehyde. PS Polystyrene. CMC Carboxymethyl cellulose. PSU Polysulphone. CN Cellulose nitrate. PTFE Polytetrafluoroethylene. CP Cellulose propionate. PUR Polyurethane. CTA Cellulose triacetate. PVAC Polyvinyl acetate. EC Ethyl cellulose. PVAL Polyvinyl alcohol. E/EA Ethylene/ethylene acrylate. PVB Polyvinyl butyral. E/MA Ethylene/methacrylic acid. PVC Polyvinyl chloride. EP Epoxide or epoxy. PVDC Polyvinylidene chloride. EIP Ethylene/propylene. PVDF Polyvinylidene fluoride. EPDM Ethylene/propylene/diene. PVF Polyvinyl fluoride. EITFE Ethylene/tetrafluoroethylene. PVFM Polyvinyl formal. EVAC Ethylene/vinyl acetate. PVK Polyvinylcarbazole. EVAL Ethylene/vinyl alcohol. PVP Polyvinylpyrrolidone. FEP Perfluoro(ethylene/propylene): SAN Styrene acrylonitrile. tetrafluoroethylenelhexafluoropropylene. SIB Styrenelbutadiene. FF Furane-formaldehyde.