Download Author Version (PDF)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemical Innovation Technologies to Make Processes and Products More Sustainable
United States Government Accountability Office Center for Science, Technology, and Engineering Natural Resources and Environment Report to Congressional Requesters February 2018 TECHNOLOGY ASSESSMENT Chemical Innovation Technologies to Make Processes and Products More Sustainable GAO-18-307 The cover image displays a word cloud generated from the transcript of the meeting we convened with 24 experts in the field of sustainable chemistry. The size of the words in the cloud corresponds to the frequency with which each word appeared in the transcript. In most cases, similar words—such as singular and plural versions of the same word— were combined into a single term. Words that were unrelated to the topic of sustainable chemistry were removed. The images around the periphery are stylized representations of chemical molecules that seek to illustrate a new conceptual framework, whereby molecules can be transformed to provide better performance; however, they are not intended to represent specific chemical compounds. TECHNOLOGY ASSESSMENT Highlights of GAO-18-307, a report to congressional requesters Chemical Innovation February 2018 Technologies to Make Processes and Products More Sustainable Why GAO did this study What GAO found Chemistry contributes to virtually every Stakeholders lack agreement on how to define sustainable chemistry and how to aspect of modern life and the chemical measure or assess the sustainability of chemical processes and products; these industry supports more than 25 percent differences hinder the development and adoption of more sustainable chemistry of the gross domestic product of the technologies. However, based on a review of the literature and stakeholder United States. While these are positive interviews, GAO identified several common themes underlying what sustainable contributions, chemical production can chemistry strives to achieve, including: have negative health and environmental · improve the efficiency with which natural resources—including energy, consequences. -

The Vibration Spectra of Hydrazoic Acid, Methyl Azide, and Methyl Isocyanate
MAY, 1940 JOURNAL OF CHEMICAL PHYSICS VOLUME 8 The Vibration Spectra of Hydrazoic Acid, Methyl Azide, and Methyl Isocyanate The Thermodynamic Functions of Hydrazoic Acid EUGENE H. EYSTER,* Department of Physics, University of Michigan, Ann Arbor, Michigan AND R. H. GILLETTE, Department of Chemistry, University of Michigan, Ann Arbor, Michigan (Received February 5, 1940) The vibration spectra of hydrazoic acid, methyl azide, and methyl isocyanate have been investigated in the spectral region between 2 and 20~. Correlation of the spectra of these struc turally similar molecules has made possible the determination of all the fundamental fre quencies of hydrazoic acid and all but the methyl torsion frequency in the methyl compounds. From these fundamental frequencies and the known rotational constants, the usual thermo dynamic functions of hydrazoic acid have been calculated to the harmonic oscillator-rigid ro tator approximation. Equilibrium constants for some characteristic reactions have also been obtained. HE structures of hydrazoic acidl and of all but the methyl torsion oscillations in the T methyl azide2 have now been determined methyl compounds. From these results and the by physical methods, and can be readily inter rotational constants of hydrazoic acid,l it has preted in the light of the modern structural been possible to calculate the thermodynamic theory. The vibration spectrum of the acid has functions of the acid, and to conclude that the also been investigated by Davies,3 but he was azide and isocyanate groups are structurally unable to decide with certainty the magnitude of similar. the two lowest frequencies. Since these two EXPERIMENTAL vibrations are confined essentially to the azide group, it was thought that a comparison of the Hydrazoic acid was prepared by the method of spectra of hydrazoic acid and methyl azide would Dennis and Isham5 from recrystallized sodium aid in 8eciding which of the low'frequency bands azide. -

ACS Guide to Scholarly Com
1.3 Communicating Safety Information Samuella Sigmann Visit the full ACS Guide to Scholarly Communication 1.3.1 Introduction In 2011, the U.S. Chemical Safety Board (CSB) issued its first investigation of a chemical accident at an academic research institution. The incident involved the sudden detonation of a nickel hydrazine perchlorate (NHP) derivative that resulted in severe and permanent injury to a fifth-year chemistry graduate student at Texas Tech University (TTU). Student researchers were involved in a multi-institutional research project to characterize new potentially energetic materials. They decided to scale up their synthesis by two orders of magnitude to provide enough sample so that all testing could be performed on one batch but neglected to assess safety ramifications. The final CSB report for the TTU incident found in both this case and prior incidents in the same research area that (1): . No formal hazard evaluation and risk assessment had been completed to char-acterize the potential danger of the research activity and to plan for the worst-case scenario. The student sought peer advice. No policy was in place at the laboratory, department, or university level to prompt a student to seek PI advice or evaluation of experimental activities. The investigation clearly indicates that the students did not have the information they needed to prepare themselves for the increased risk posed by their decision to scale up. The CSB primarily investigates and reports on large scale incidents in the chemical and related industries where hazard evaluation and risk assessment are well established to facilitate improved safety management across the industry. -

Ultrafast 25-Fs Relaxation in Highly Excited States of Methyl Azide
Ultrafast 25-fs relaxation in highly excited states of PNAS PLUS methyl azide mediated by strong nonadiabatic coupling William K. Petersa,b,1, David E. Coucha,b, Benoit Mignoletc, Xuetao Shid, Quynh L. Nguyena,b, Ryan C. Fortenberrye, H. Bernhard Schlegeld, Françoise Remaclec, Henry C. Kapteyna,b, Margaret M. Murnanea,b,1, and Wen Lid aJILA, University of Colorado, Boulder, CO 80309; bDepartment of Physics, University of Colorado, Boulder, CO 80309; cTheoretical Physical Chemistry, Unité de Recherche Molecular Systems (UR MOLSYS) University of Liège, B4000 Liège, Belgium; dDepartment of Chemistry, Wayne State University, Detroit, MI 48202; and eDepartment of Chemistry and Biochemistry, Georgia Southern University, Statesboro, GA 30460 Contributed by Margaret M. Murnane, October 14, 2017 (sent for review July 16, 2017; reviewed by Elliot Bernstein and Arthur Suits) Highly excited electronic states are challenging to explore exper- When molecules are excited by vacuum-UV (VUV) light in imentally and theoretically—due to the large density of states and the 7- to 10-eV range, just below the ionization potential, one or the fact that small structural changes lead to large changes in several excited states can be accessed depending on the band- electronic character with associated strong nonadiabatic dynamics. width of the pulse. In this energy range, there is typically a dense They can play a key role in astrophysical and ionospheric chemis- manifold of valence and Rydberg states that are often strongly try, as well as the detonation chemistry of high-energy density coupled, each with an associated vibrational structure (9). As a materials. Here, we implement ultrafast vacuum-UV (VUV)-driven result, VUV molecular absorption spectra typically contain electron–ion coincidence imaging spectroscopy to directly probe quasicontinuous but structured bands with overlapping sharp the reaction pathways of highly excited states of energetic mole- peaks, indicating the wide variety of timescales and states in- — cules in this case, methyl azide. -

Investigations Towards the Design, Synthesis and Application of New Sulfur-Based Transfer Reagents
Investigations towards the design, synthesis and application of new sulfur-based transfer reagents Dissertation zur Erlangung des mathematisch-naturwissenschaftlichen Doktorgrades „Doctor rerum naturalium“ der Georg-August-Universität Göttingen im Promotionsprogramm der Georg-August University School of Science (GAUSS) vorgelegt von Bernd Waldecker aus Emden Göttingen, 2019 Betreuungsausschuss: Prof. Dr. M. Alcarazo (Institut für Organische und Biomolekulare Chemie, Tammannstr. 2, 37077 Göttingen) Prof. Dr. L. Ackermann (Institut für Organische und Biomolekulare Chemie, Tammannstr. 2, 37077 Göttingen) Mitglieder der Prüfungskommission: Referent: Prof. Dr. M. Alcarazo (Institut für Organische und Biomolekulare Chemie, Tammannstr. 2, 37077 Göttingen) Korreferent: Prof. Dr. L. Ackermann (Institut für Organische und Biomolekulare Chemie, Tammannstr. 2, 37077 Göttingen) Weitere Mitglieder der Prüfungskommission: Prof. Dr. D. Stalke (Institut für Anorganische Chemie, Tammannstr. 4, 37077 Göttingen) Dr. S. Das (Institut für Organische und Biomolekulare Chemie, Tammannstr. 2, 37077 Göttingen) Dr. F. Thomas (Institut für Organische und Biomolekulare Chemie, Tammannstr. 2, 37077 Göttingen) Dr. M. Hansmann (Institut für Organische und Biomolekulare Chemie, Tammannstr. 2, 37077 Göttingen) Tag der mündlichen Prüfung: 02.05.2019 Die vorliegende Arbeit entstand unter Anleitung von Herrn. Prof. Dr. Manuel Alcarazo in der Zeit von Juli 2015 bis Dezember 2015 am Max‐Planck‐Institut für Kohlenforschung in Mülheim an der Ruhr und in der Zeit von Januar 2016 bis Dezember 2018 an der Georg- August-Universität zu Göttingen. Teile dieser Arbeit wurden bereits veröffentlicht: J. Peña, G. Talavera, B. Waldecker, M. Alcarazo, Chem. Eur. J. 2017, 23, 75-78. B. Waldecker, F. Kraft, C. Golz, M. Alcarazo, Angew. Chem. Int. Ed. 2018, 57, 12538-12542. Ein Patent zu den in der Arbeit synthetisierten Verbindungen und zu deren synthetischer Anwendbarkeit wurde von der Universität Göttingen eingereicht (DE102018211606.7). -

PUV3402, PIR3502 and PFO3372 Process Photometers Applications, Technology and Data Process Photometers PUV3402 and PIR3502
ABB MultiwaveTM process photometers ABB Analytical - PUV3402, PIR3502 and PFO3372 process photometers Applications, technology and data Process photometers PUV3402 and PIR3502 Principle of operation Advantages of the design The PUV3402 and PIR3502 process photometers optical path The PUV3402 and PIR3502 process photometers use a fixed consists of an IR or UV source, a brushless chopper motor, wavelength filter, single beam, multichannel principle. This a multichannel filter wheel, lenses, cell with windows, and a design concept offers several advantages: solid state IR or UV detector. (See Optical Schematic). – Produces a simple mechanical design that promotes easier service and maintenance. The brushless chopper motor rotates the multichannel filter – Compensates for obstruction of cell windows. wheel reference and measure filters, alternately and – Compensates for source and detector aging. continuously, into and out of the optical path. The lenses, – Permits the sample cell to be isolated from the electronics. L1 through L4, focus and collimate the source radiation – Enable multicomponent analysis. through the sample cell path to the detector. As illustrated by the optical schematic, the simple mechanical On a multichannel photometer, a reference wavelength is design has a single optical path, from the source directly to chosen where the stream components have little or no the detector. The source energy is focused and collimated absorbance. The measure wavelength or wavelengths are straight to the detector. It does not require optical mirrors or chosen where the measured components have absorbance. internal reflections. The microprocessor will then use matrix algebra and apply the proper response factor to each filter to eliminate interferences on the desired component and convert the absorbance to a component concentration. -

The Synthesis and Characterisation of Halogen and Nitro Phenyl Azide
Dissertation zur Erlangung des Dokorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilans-Universität München The synthesis and characterisation of halogen and nitro phenyl azide derivatives as highly energetic materials. von David Adam aus Glasgow , Schottland 2001 Index Acknowledgements 1 Abstract 3 1. Introduction 4 1.1 Azides 4 1.2 Nitro compounds 13 1.3 Nitrocarbons 16 1.4 Explosives 18 1.5 Theoretical Calculations 20 1.6 Pentazoles 23 2 Halogen substitued phenyl azides 27 2.1 2,4,6-tribromophenyl azide 27 2.2 2,4,6-trichlorophenyl azide 35 2.3 2,5,6-trichlorophenyl azide 42 2.4 2,4-dibromophenyl azide 43 2.5 2,4-dichlorophenyl azide 44 2.6 2,4,6-trifluorophenyl azide 44 2.7 2,3,4,5,6-pentafluorophenyl azide 47 2.8 2,6-diiodo-4-nitrophenyl azide 50 2.9 2,3,4,5,6-pentachlorophenyl azide 59 3. Nitro compounds 65 3.1 o-nitrophenyl azide 65 3.2 m-nitrophenyl azide 68 3.3 2,4-dinitrophenyl azide 70 3.4 2,4,6-trintrophenyl azide 72 3.5 1,3,5-triazido-2,4-dinitrobenzene 94 3.6 1,3,5-triazido-2,4,6-trinitrobenzene 100 3.7 hexakis (azidomethyl) benzene. 106 3.8 hexakis (methylnitrate) benzene 108 4. Decomposition and explosion experiments 111 4.1 Thermal decomposition of TNMA , DNTA and TNTA 111 4.2 Computational Aspects 118 4.3 Thermal decomposition of halogen substituted phenyl azides 121 4.4 Thermal decomposition of hexakis (azidomethyl) benzene 122 4.5 Drophammer Tests of TNMA, DNTA and TNTA 125 4.6 Calculation of the detonation velocity from molecular formula and structure for TNMA , DNTA and TNTA 133 4.7 Calculation of the detonation velocity from molecular formula and structure for a series of azido and nitro substituted benzenes 135 5 Calculation of compounds of the type C6 (NO2)6-n (N3)n 138 5.1 Structural discussion 144 5.2 Molecular structures 145 6. -
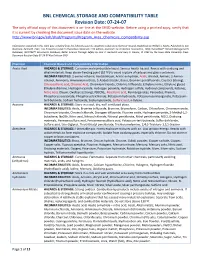
BNL CHEMICAL STORAGE and COMPATIBILITY TABLE Revision Date: 07-24-07 the Only Official Copy of This Document Is On-Line at the SHSD Website
BNL CHEMICAL STORAGE AND COMPATIBILITY TABLE Revision Date: 07-24-07 The only official copy of this document is on-line at the SHSD website. Before using a printed copy, verify that it is current by checking the document issue date on the website. http://www.bnl.gov/esh/shsd/Programs/Program_Area_Chemicals_Compatibility.asp Information contained in this table was compiled from the following sources: Academic Laboratory Chemical Hazards Guidebook by William J. Mahn, Published by Van Nostrand, Reinhold, 1991; Fire Protection Guide to Hazardous Materials 11th edition, National Fire Protection Association, 1994; Hazardtext® Hazard Managements Database; INFOTEXT® Documents Database; Better Science Through Safety by Jack A. Gerlovich and Gary E. Downs, © 1981 by the Iowa State University Press. Document Revision Date 07-24-07 Ken Erickson CHO Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 ºF) to avoid rupture of carboys and glass containers. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino- ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers -

I. Electrophilic Reactions of .E -Toluenesulfonyl Azide Ii
I. ELECTROPHILIC REACTIONS OF .E_-TOLUENESULFONYL AZIDE 15 13 II. N AND c NUCLEAR MAGNETIC STUDIES OF ARYLDIAZONIUM COMPOUNDS EFFECT OF SUBSTITUENT~ SOLVENT AND 18-CROWN-6 Thesis by Carla Jutta Casewit In Partial Fulfillment of the Requirements for the Degree of .Doctor of Philosophy California Institute of Technology Pasadena, California 1981 (Submitted November 1, 1980) ; ; To Tante Renate iii ACKNOWLEDGMENTS I thank John D. Roberts for his guidance, encouragement and most of all his imagination and honesty. I thank the members of the Roberts group, past and present, for the many long discussions and excursions. I am especially grateful to com rades in arms Mike Nee, Nancy Becker, and Nina Gonnella. I am indebted to Amy Abe for her generous help in getting started. I am grateful to David Horowitz for the two years of unmatched wit and humor. I thank Bill Goddard for giving me the opportunity to "go where my scientific curiousity drove me", and the members of his group for their warm and lively companionship. I owe much to the faculty of the University of Colorado at Denver. I am especially grateful to Bob and Lennie Damrauer for their encourage ment and friendship then and now. IBM and Caltech Fellowships are gratefully acknowledged. Most of all I thank my family for their support and interest, and Tony for his insight, encouragement and love. iv ABSTRACT PART I ELECTROPHILIC REACTIONS OF .E_-TOLUENESULFONYL AZIDE Section 1. Review of Electrophilic Reactions of £.-Toluenesulfonyl Azide The electrophilic reactions of .E_-toluenesulfonyl azide are reviewed using the principle of hard and soft acids and bases (HSAB). -

Report-Of Committee on Chemicals and Explosives
448 REPORT OF COMMITTEE ON CHEMICALS AND EXPLOSIVES CE-1 Report-of Committee on Chemicals and Explosives Correlating Committee Dr. Robert W. Van Dolah, Chairman, Pittsburgh Mining and Safety Research Center, Bureau of Mines, U.S. Department of the Interior, 4800 Forbes Ave., Pittsburgh, PA 15213 Chester I. Babeock,~ Secretary, National Pire Protection Assn., 470 Atlantic Ave., Boston, MA 02210 W. H. Doyle, Simsbury, CT ilenry T. Rlttman, Institute of Makers of •, Thomas E. Duke, Fire Prevention & Engi- Explosives neering Bureau of Texas Richard F. Schwab, Allied Chemical Corp. Dr. Richard Y. Le Vine, Olin Corp. tNonvoting. Sectional Committee on Electrical Equipment in Chemical Atmospheres Dr. Richard Y. Le Vine, Chairman, Olin Corp., 120 Long Ridge Rd., Stamford, CT 06904 Chester I. Babcock,~ Secretary, National Fire Protection Association, 470 Atlantic Ave., Boston, MA 02210 L. J. Hall. Panel No. 14, National Electrical R. F. Schwab, Morristown, NJ Code Committee W. A. Short, National Electrical-Manu- • Robert P. llowell, American Petroleu~i In" facturers Assn. stitute George O. Hunt, Jr., Manufacturing Chem- Alternates. ists' Assn. Elton L. Lltehfleld, Pittsburgh, PA F. D. Alroth. (Alternate to P. J. Schram) Frederick L. Maltby, Instrument Society W. Calder (Alternate to F. L. Maltby) of America W. H. Levers (Alternate to Robert P. C. E. Miller, Norwood, MA Howell) " Frank E. Rademacher, Chicago, IL J. Rennle (Alternate to C. E. Miller) John E. Rogerson. Cincinnati, OH Thomas S. Staron, (Alternate to Frank E. P. J. Schram, Chicago, IL Rademaehcr) tNonvoting 449 CE-2 EXPLANATION OF REPORT Sectional Committee on llazardous Chemical Reactions R. F. Schwab, Chairman, Allied Chemical Corp., P.O. -

Peroxide-Forming Chemicals
Peroxide-Forming Chemicals Overview Peroxide-forming chemicals are a class of compounds that have the ability to form shock-sensitive explosive peroxide crystals. Many of the organic solvents commonly used in laboratories have the potential to form explosive peroxide crystals. Diethyl ether and tetrahydrofuran are two of the more common peroxide-forming chemicals used today. Therefore, it is extremely important that this procedure be followed regarding the identification, handling, storage, and disposal of peroxide-forming chemicals. Peroxide Formation Under normal storage conditions the materials listed in this document have the potential to generate and accumulate peroxide crystal formations, which may violently detonate when subjected to thermal or mechanical shock. Peroxide-forming chemicals react with oxygen – even at low concentrations – to form peroxy compounds. The risk associated with peroxide formation increases if the peroxide crystallizes or becomes concentrated by evaporation or distillation. Factors that affect rate of peroxide formation include exposure to air, light and heat, moisture, and contamination from metals. Manufacturers may add an inhibitor to peroxide forming chemicals to counter peroxide formation. For many peroxide-forming solvents, butylated hydroxy toluene (BHT) is commonly added. BHT ‘scavenges’ oxygen in the solvent and prevents it from reacting with the solvent to form peroxides. Over time, BHT or other inhibitor in the solvent can become exhausted allowing peroxides to form. Distilling the solvent can completely remove the BHT and make the solvent immediately susceptible to peroxide formation. Peroxide crystals may form on the container plug or the threads of the lid and detonate when the lid is twisted. Do not open a liquid organic peroxide or peroxide-forming chemical if crystals or a precipitate are present. -

Department of Labor
Vol. 79 Friday, No. 197 October 10, 2014 Part II Department of Labor Occupational Safety and Health Administration 29 CFR Parts 1910, 1915, 1917, et al. Chemical Management and Permissible Exposure Limits (PELs); Proposed Rule VerDate Sep<11>2014 17:41 Oct 09, 2014 Jkt 235001 PO 00000 Frm 00001 Fmt 4717 Sfmt 4717 E:\FR\FM\10OCP2.SGM 10OCP2 mstockstill on DSK4VPTVN1PROD with PROPOSALS2 61384 Federal Register / Vol. 79, No. 197 / Friday, October 10, 2014 / Proposed Rules DEPARTMENT OF LABOR faxed to the OSHA Docket Office at Docket: To read or download (202) 693–1648. submissions or other material in the Occupational Safety and Health Mail, hand delivery, express mail, or docket go to: www.regulations.gov or the Administration messenger or courier service: Copies OSHA Docket Office at the address must be submitted in triplicate (3) to the above. All documents in the docket are 29 CFR Parts 1910, 1915, 1917, 1918, OSHA Docket Office, Docket No. listed in the index; however, some and 1926 OSHA–2012–0023, U.S. Department of information (e.g. copyrighted materials) Labor, Room N–2625, 200 Constitution is not publicly available to read or [Docket No. OSHA 2012–0023] Avenue NW., Washington, DC 20210. download through the Web site. All submissions, including copyrighted RIN 1218–AC74 Deliveries (hand, express mail, messenger, and courier service) are material, are available for inspection Chemical Management and accepted during the Department of and copying at the OSHA Docket Office. Permissible Exposure Limits (PELs) Labor and Docket Office’s normal FOR FURTHER INFORMATION CONTACT: business hours, 8:15 a.m.