Mutation Analysis and Embryonic Expression of the HLXB9 Currarino Syndrome Gene D
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Genetic Heterogeneity of Brachydactyly Type A1: Identifying the Molecular Pathways
The genetic heterogeneity of brachydactyly type A1: Identifying the molecular pathways Lemuel Jean Racacho Thesis submitted to the Faculty of Graduate Studies and Postdoctoral Studies in partial fulfillment of the requirements for the Doctorate in Philosophy degree in Biochemistry Specialization in Human and Molecular Genetics Department of Biochemistry, Microbiology and Immunology Faculty of Medicine University of Ottawa © Lemuel Jean Racacho, Ottawa, Canada, 2015 Abstract Brachydactyly type A1 (BDA1) is a rare autosomal dominant trait characterized by the shortening of the middle phalanges of digits 2-5 and of the proximal phalange of digit 1 in both hands and feet. Many of the brachymesophalangies including BDA1 have been associated with genetic perturbations along the BMP-SMAD signaling pathway. The goal of this thesis is to identify the molecular pathways that are associated with the BDA1 phenotype through the genetic assessment of BDA1-affected families. We identified four missense mutations that are clustered with other reported BDA1 mutations in the central region of the N-terminal signaling peptide of IHH. We also identified a missense mutation in GDF5 cosegregating with a semi-dominant form of BDA1. In two families we reported two novel BDA1-associated sequence variants in BMPR1B, the gene which codes for the receptor of GDF5. In 2002, we reported a BDA1 trait linked to chromosome 5p13.3 in a Canadian kindred (BDA1B; MIM %607004) but we did not discover a BDA1-causal variant in any of the protein coding genes within the 2.8 Mb critical region. To provide a higher sensitivity of detection, we performed a targeted enrichment of the BDA1B locus followed by high-throughput sequencing. -

Pushing the Limits of Prenatal Ultrasound: a Case of Dorsal Dermal Sinus Associated with an Overt Arnold–Chiari Malformation and a 3Q Duplication
reproductive medicine Case Report Pushing the Limits of Prenatal Ultrasound: A Case of Dorsal Dermal Sinus Associated with an Overt Arnold–Chiari Malformation and a 3q Duplication Olivier Leroij 1, Lennart Van der Veeken 2,*, Bettina Blaumeiser 3 and Katrien Janssens 3 1 Faculty of Medicine, University of Antwerp, 2610 Wilrijk, Belgium; [email protected] 2 Department of Obstetrics and Gynaecology, University Hospital Antwerp, 2650 Edegem, Belgium 3 Department of Medical Genetics, University Hospital and University of Antwerp, 2650 Edegem, Belgium; [email protected] (B.B.); [email protected] (K.J.) * Correspondence: [email protected] Abstract: We present a case of a fetus with cranial abnormalities typical of open spina bifida but with an intact spine shown on both ultrasound and fetal MRI. Expert ultrasound examination revealed a very small tract between the spine and the skin, and a postmortem examination confirmed the diagnosis of a dorsal dermal sinus. Genetic analysis found a mosaic 3q23q27 duplication in the form of a marker chromosome. This case emphasizes that meticulous prenatal ultrasound examination has the potential to diagnose even closed subtypes of neural tube defects. Furthermore, with cerebral anomalies suggesting a spina bifida, other imaging techniques together with genetic tests and measurement of alpha-fetoprotein in the amniotic fluid should be performed. Citation: Leroij, O.; Van der Veeken, Keywords: dorsal dermal sinus; Arnold–Chiari anomaly; 3q23q27 duplication; mosaic; marker chro- L.; Blaumeiser, B.; Janssens, K. mosome Pushing the Limits of Prenatal Ultrasound: A Case of Dorsal Dermal Sinus Associated with an Overt Arnold–Chiari Malformation and a 3q 1. -

Prevalence and Incidence of Rare Diseases: Bibliographic Data
Number 1 | January 2019 Prevalence and incidence of rare diseases: Bibliographic data Prevalence, incidence or number of published cases listed by diseases (in alphabetical order) www.orpha.net www.orphadata.org If a range of national data is available, the average is Methodology calculated to estimate the worldwide or European prevalence or incidence. When a range of data sources is available, the most Orphanet carries out a systematic survey of literature in recent data source that meets a certain number of quality order to estimate the prevalence and incidence of rare criteria is favoured (registries, meta-analyses, diseases. This study aims to collect new data regarding population-based studies, large cohorts studies). point prevalence, birth prevalence and incidence, and to update already published data according to new For congenital diseases, the prevalence is estimated, so scientific studies or other available data. that: Prevalence = birth prevalence x (patient life This data is presented in the following reports published expectancy/general population life expectancy). biannually: When only incidence data is documented, the prevalence is estimated when possible, so that : • Prevalence, incidence or number of published cases listed by diseases (in alphabetical order); Prevalence = incidence x disease mean duration. • Diseases listed by decreasing prevalence, incidence When neither prevalence nor incidence data is available, or number of published cases; which is the case for very rare diseases, the number of cases or families documented in the medical literature is Data collection provided. A number of different sources are used : Limitations of the study • Registries (RARECARE, EUROCAT, etc) ; The prevalence and incidence data presented in this report are only estimations and cannot be considered to • National/international health institutes and agencies be absolutely correct. -

Orphanet Report Series Rare Diseases Collection
Marche des Maladies Rares – Alliance Maladies Rares Orphanet Report Series Rare Diseases collection DecemberOctober 2013 2009 List of rare diseases and synonyms Listed in alphabetical order www.orpha.net 20102206 Rare diseases listed in alphabetical order ORPHA ORPHA ORPHA Disease name Disease name Disease name Number Number Number 289157 1-alpha-hydroxylase deficiency 309127 3-hydroxyacyl-CoA dehydrogenase 228384 5q14.3 microdeletion syndrome deficiency 293948 1p21.3 microdeletion syndrome 314655 5q31.3 microdeletion syndrome 939 3-hydroxyisobutyric aciduria 1606 1p36 deletion syndrome 228415 5q35 microduplication syndrome 2616 3M syndrome 250989 1q21.1 microdeletion syndrome 96125 6p subtelomeric deletion syndrome 2616 3-M syndrome 250994 1q21.1 microduplication syndrome 251046 6p22 microdeletion syndrome 293843 3MC syndrome 250999 1q41q42 microdeletion syndrome 96125 6p25 microdeletion syndrome 6 3-methylcrotonylglycinuria 250999 1q41-q42 microdeletion syndrome 99135 6-phosphogluconate dehydrogenase 67046 3-methylglutaconic aciduria type 1 deficiency 238769 1q44 microdeletion syndrome 111 3-methylglutaconic aciduria type 2 13 6-pyruvoyl-tetrahydropterin synthase 976 2,8 dihydroxyadenine urolithiasis deficiency 67047 3-methylglutaconic aciduria type 3 869 2A syndrome 75857 6q terminal deletion 67048 3-methylglutaconic aciduria type 4 79154 2-aminoadipic 2-oxoadipic aciduria 171829 6q16 deletion syndrome 66634 3-methylglutaconic aciduria type 5 19 2-hydroxyglutaric acidemia 251056 6q25 microdeletion syndrome 352328 3-methylglutaconic -

Spectrum of Mutations and Genotype ± Phenotype Analysis in Currarino Syndrome
European Journal of Human Genetics (2001) 9, 599 ± 605 ã 2001 Nature Publishing Group All rights reserved 1018-4813/01 $15.00 www.nature.com/ejhg ARTICLE Spectrum of mutations and genotype ± phenotype analysis in Currarino syndrome Joachim KoÈchling1, Mohsen Karbasiyan2 and Andre Reis*,2,3 1Department of Pediatric Oncology/Hematology, ChariteÂ, Humboldt University, Berlin, Germany; 2Institute of Human Genetics, ChariteÂ, Humboldt University, Berlin, Germany; 3Institute of Human Genetics, Friedrich- Alexander University Erlangen-NuÈrnberg, Erlangen, Germany The triad of a presacral tumour, sacral agenesis and anorectal malformation constitutes the Currarino syndrome which is caused by dorsal-ventral patterning defects during embryonic development. The syndrome occurs in the majority of patients as an autosomal dominant trait associated with mutations in the homeobox gene HLXB9 which encodes the nuclear protein HB9. However, genotype ± phenotype analyses have been performed only in a few families and there are no reports about the specific impact of HLXB9 mutations on HB9 function. We performed a mutational analysis in 72 individuals from nine families with Currarino syndrome. We identified a total of five HLXB9 mutations, four novel and one known mutation, in four out of four families and one out of five sporadic cases. Highly variable phenotypes and a low penetrance with half of all carriers being clinically asymptomatic were found in three families, whereas affected members of one family showed almost identical phenotypes. However, an obvious genotype ± phenotype correlation was not found. While HLXB9 mutations were diagnosed in 23 patients, no mutation or microdeletion was detected in four sporadic patients with Currarino syndrome. The distribution pattern of here and previously reported HLXB9 mutations indicates mutational predilection sites within exon 1 and the homeobox. -

Test Catalogue August 2019
Test Catalogue August 2019 www.centogene.com/catalogue Table of Contents CENTOGENE CLINICAL DIAGNOSTIC PRODUCTS AND SERVICES › Whole Exome Testing 4 › Whole Genome Testing 5 › Genome wide CNV Analysis 5 › Somatic Mutation Analyses 5 › Biomarker Testing, Biochemical Testing 6 › Prenatal Testing 7 › Additional Services 7 › Metabolic Diseases 9 - 21 › Neurological Diseases 23 - 47 › Ophthalmological Diseases 49 - 55 › Ear, Nose and Throat Diseases 57 - 61 › Bone, Skin and Immune Diseases 63 - 73 › Cardiological Diseases 75 - 79 › Vascular Diseases 81 - 82 › Liver, Kidney and Endocrinological Diseases 83 - 89 › Reproductive Genetics 91 › Haematological Diseases 93 - 96 › Malformation and/or Retardation Syndromes 97 - 107 › Oncogenetics 109 - 113 ® › CentoXome - Sequencing targeting exonic regions of ~20.000 genes Test Test name Description code CentoXome® Solo Medical interpretation/report of WES findings for index 50029 CentoXome® Solo - Variants Raw data; fastQ, BAM, Vcf files along with variant annotated file in xls format for index 50028 CentoXome® Solo - with CNV Medical interpretation/report of WES including CNV findings for index 50103 Medical interpretation/report of WES in index, package including genome wide analyses of structural/ CentoXome® Solo - with sWGS 50104 large CNVs through sWGS Medical interpretation/report of WES in index, package including genome wide analyses of structural/ CentoXome® Solo - with aCGH 750k 50122 large CNVs through 750k microarray Medical interpretation/report of WES in index, package including genome -

Περιεχόμενα: I: GENERAL CONCEPTS 1. Human
Περιεχόμενα: I: GENERAL CONCEPTS 1. Human Malformations and Their Genetic Basis CHARLES J. EPSTEIN 2. Principles of Differentiation and Morphogenesis scott f. gilbert and ritva rice 3. Model Organisms in the Study of Development and Disease ethan bier and william mcginnis 4. Human Genomics and Human Development Bob Nussbaum II: Patterns of Development 5. Development of Left-Right Asymmetry Hiroshi Hamada 6. Neural Crest Formation and Craniofacial Development Kurt A. Engleka and Jonathan A. Epstein 7. Development of the Nervous System JOHN L. R. RUBENSTEIN AND LUIS PUELLES 8. Development of the Eye David C. Beebe 9. Development of the Ear Donna M. Fekete 10. Molecular Regulation of Cardiogenesis Deepak Srivastava and Joseph T. C. Shieh 11. Update on the Development of the Vascular System and Its Sporadic Disorders M. Michael Cohen Jr 12. MUSCLE AND SOMITE DEVELOPMENT Douglas Anderson and Alan Rawls 13. The Development of Bone and Cartilage shunichi murakami, haruhiko akiyama, And benoit de crombrugghe 14. LIMB DEVELOPMENT MalteSpielmann and Sigmar Stricker 15. The Sex Determination Pathway PETER J. eLLIS and robert p. erickson 16. Development of the Kidney Kevin T. Bush, Mita M. Shah, Dylan L. Steer, Derina E. Sweeney, and Sanjay K. Nigam 17. DEVELOPMENT OF THE ENDODERMAL DERIVATIVES IN LUNG, LIVER, PANCREAS, AND GUT Ben Z. Stanger, 18. Development of Epidermal Appendages: Teeth and Hair ATSUSHI OHAZAMA AND PAUL T. SHARPE III: Defined Core Developmental Pathways Linked to Cilia Part A: Ciliary Functions: Genesis, Transport, and Reabsorbtion 19. Primary Ciliary Dyskinesia (Kartagener's Syndrome) MICHAL WITT AND ZUZANNA BUKOWY-BIERY??O 20. The Molecular Basis of Joubert Syndrome and Related Disorders Jeong Ho Lee and Joseph G. -

2 Genetics of Anorectal Malformations
17 2 Genetics of Anorectal Malformations Giuseppe Martucciello Contents blastogenesis often involve two or more progenitor fields. This fact may explain the cause of subgroups 2.1 Introduction . 17 of ARM that form part of complex phenotypes due 2.2 Non-syndromic ARM . 17 to developmental field defects. These complex phe- 2.3 ARM Associated with Other Systemic notypes are considered as end results of pleiotropic Malformations . 18 effects of single causal events that might be chromo- 2.4 Aetiological Classification . 20 somal, monogenic or even teratogenic [72]. 2.4.1 Chromosomal Anomalies Classifying ARM from the genetic point of view is Associated with ARM . 20 2.4.2 Down Syndrome . 20 not easy since they present different forms that are be- 2.4.3 Cat-Eye Syndrome . 20 lieved to be influenced by different factors such as sex 2.4.4 Genetic Syndromes Associated and associated anomalies. ARM can be the only path- with ARM . 21 ological finding (non-syndromic) or as part of a more 2.4.4.1 Townes-Brocks Syndrome . 21 complex phenotype (syndromic), and may occur in a 2.4.4.2 FG Syndrome . 21 single affected individual (sporadic) or in more than 2.4.4.3 Pallister-Hall Syndrome . 21 one individual in the same family (familial) with dif- 2.4.4.4 VACTERL Association (VATER) . 22 ferent modes of inheritance. There are gender differ- 2.4.4.5 Sirenomelia . 22 ences, with remarkably higher preponderance in boys 2.4.4.6 Caudal Regression Syndrome . 23 for more complex ARM forms, while the less severe 2.4.4.7 Currarino Syndrome . -

Chromosomal Microarray, Postnatal, Clarisure® Oligo-SNP
Diagnostic Services Pediatrics Test Summary Chromosomal Microarray, Postnatal, ClariSure® Oligo-SNP The American College of Medical Genetics (ACMG) Test Code: 16478(X) recommends CMA testing as a first-line genetic test for developmental delay, intellectual disability, ASDs, and Specimen Requirements: 10 mL room-temperature whole multiple congenital anomalies.2,4 This recommendation blood (sodium-heparin, green-top tube); 5 mL minimum is based, in part, on a literature review that included over 21,000 patients with developmental delay/intellectual CPT Code*: 81229 disability, ASDs, or multiple congenital anomalies. The diagnostic yield was 15% to 20% for CMA testing versus ~3% for G-banded karyotyping and ~6% for subtelomeric Clinical Use fluorescence in situ hybridization (FISH) in combination • Determine the genetic etiology of developmental with G-banded karyotyping.5 In individuals with complex delay, intellectual disability, autism spectrum disorders ASDs, CMA testing can result in a diagnostic yield of over (ASDs; pervasive developmental disorders), and 25%.2 ACMG still considers karyotyping a first-line test multiple congenital anomalies when patients are suspected of having a recognizable • Confirm or exclude the diagnosis of known chromosomal syndrome such as trisomy 21 or 18, Turner chromosomal syndromes syndrome, or Klinefelter syndrome.2,4 • Further define ambiguities arising from cytogenetic The oligonucleotide-single nucleotide polymorphism or FISH studies (oligo-SNP) array contains over 2.6 million probes and • Assist in clinical management and genetic counseling covers regions of known and likely CNVs. It can confirm the diagnosis of suspected disorders associated with known Clinical Background chromosomal syndromes and is especially well suited Global developmental delay, intellectual disability (mental for determining the genetic cause of less well-described retardation), ASDs, and multiple congenital anomalies may disorders. -
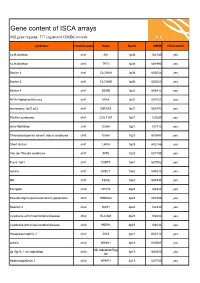
ISCA Disease List
Gene content of ISCA arrays 402 gene regions, 377 registered OMIM records syndrome chromosome Gene Band OMIM ISCA 8x60k 1p36 deletion chr1 SKI 1p36 164780 yes 1p36 deletion chr1 TP73 1p36 601990 yes Bartter 4 chr1 CLCNKA 1p36 602024 yes Bartter 3 chr1 CLCNKB 1p36 602023 yes Bartter 4 chr1 BSND 1p32 606412 yes NFIA Haploinsufficiency chr1 NFIA 1p31 600727 yes monosomy 1p31 p22 chr1 DIRAS3 1p31 605193 yes Stickler syndrome chr1 COL11A1 1p21 120280 yes atrial fibrillation chr1 GJA5 1q21 121013 yes Thrombocytopenia absent radius syndrome chr1 GJA8 1q21 600897 yes Short stature chr1 LHX4 1q25 602146 yes Van der Woude syndrome chr1 IRF6 1q32 607199 yes Fryns 1q41 chr1 DISP1 1q41 607502 yes autism chr1 DISC1 1q42 605210 yes MR chr1 TBCE 1q42 604934 yes Feingold chr2 MYCN 2q24 164840 yes Pseudovaginal perineoscrotal hypospadias chr2 SRD5A2 2p23 607306 yes Noonan 4 chr2 SOS1 2p22 182530 yes Cystinuria with mitochondrial disease chr2 SLC3A1 2p21 104614 yes Cystinuria with mitochondrial disease chr2 PREPL 2p21 104614 yes Holopresencaphly 2 chr2 SIX3 2p21 603714 yes autism chr2 NRXN1 2p16 600565 yes MicrodeletionReg 2p15p16.1 microdeletion chr2 2p15 602559 yes ion Nephronophthsis 1 chr2 NPHP1 2q13 607100 yes Holoprosencephaly 9 chr2 GLI2 2q14 165230 yes visceral heterotaxy chr2 CFC1 2q21 605194 yes Mowat-Wilson syndrome chr2 ZEB2 2q22 605802 yes autism chr2 SLC4A10 2q24 605556 yes SCN1A-related seizures chr2 SCN1A 2q24 182389 yes HYPOMYELINATION, GLOBAL CEREBRAL chr2 SLC25A12 2q31 603667 yes Split/hand foot malformation -5 chr2 DLX1 2q31 600029 yes -
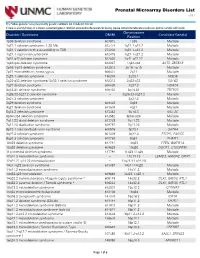
Prenatal Microarray Disorders List V19.1
Prenatal Microarray Disorders List v19.1 This "whole genome" array may identify genetic conditions not included in this list. If there is a family history of a known suspected genetic condition unrelated to the reason for testing, please contact the laboratory to discuss prior to sample submission. Chromosome Disorder / Syndrome OMIM Candidate Gene(s) Position 1p36 deletion syndrome 607872 1p36 Multiple 1q21.1 deletion syndrome, 1.35 Mb 612474 1q21.1-q21.2 Multiple 1q21.1 deletion with susceptibility to TAR 274000 1q21.1-q21.2 Multiple 1q21.1 duplication syndrome 612475 1q21.1-q21.2 Multiple 1q41-q42 deletion syndrome 612530 1q41-q42.12 Multiple 1q43-q44 deletion syndrome 612337 1q43-q44 AKT3, ZBTB18 2p16.1-p15 deletion syndrome 612513 2p16.1-p15 Multiple 2p21 microdeletion, homozygous 606407 2p21 Multiple 2q23.1 deletion syndrome 156200 2q23.1 MBD5 2q32-q33 deletion syndrome/ 2q33.1 deletion syndrome 612313 2q32-q33 SATB2 2q37 deletion syndrome 600430 2q37.3 HDAC4 3q13.31 deletion syndrome 615433 3q13.31 ZBTB20 3q26.33-3q27.2 deletion syndrome -- 3q26.33-3q27.2 Multiple 3q27.3 deletion syndrome -- 3q27.3 Multiple 3q29 deletion syndrome 609425 3q29 Multiple 4q21 deletion syndrome 613509 4q21 Multiple 5q14.3 deletion syndrome 613443 5q14.3 MEF2C 6pter-p24 deletion syndrome 612582 6pter-p24 Multiple 7q11.23 distal deletion syndrome 613729 7q11.23 Multiple 7q11.23 duplication syndrome 609757 7q11.23 Multiple 8p23.1 deletion/duplication syndrome 600576 8p23.1 GATA4 9q22.3 deletion syndrome 601309 9q22.3 PTCH1, FANCC 9q34.3 deletion syndrome -

Utviklingsavvik V02
2/1/2021 Utviklingsavvik v02 Avdeling for medisinsk genetikk Utviklingsavvik Genpanel, versjon v02 * Enkelte genomiske regioner har lav eller ingen sekvensdekning ved eksomsekvensering. Dette skyldes at de har stor likhet med andre områder i genomet, slik at spesifikk gjenkjennelse av disse områdene og påvisning av varianter i disse områdene, blir vanskelig og upålitelig. Disse genetiske regionene har vi identifisert ved å benytte USCS segmental duplication hvor områder større enn 1 kb og ≥90% likhet med andre regioner i genomet, gjenkjennes (https://genome.ucsc.edu). For noen gener ligger alle ekson i områder med segmentale duplikasjoner: ACTB, ACTG1, ASNS, ATAD3A, CA5A, CFC1, CLCNKB, CYCS, DDX11, GBA, GJA1, MSTO1, PIGC, RBM8A, RPL15, SBDS, SDHA, SHOX, SLC6A8 Vi gjør oppmerksom på at ved identifiseringav ekson oppstrøms for startkodon kan eksonnummereringen endres uten at transkript ID endres. Avdelingens websider har en full oversikt over områder som er affisert av segmentale duplikasjoner. ** Transkriptets kodende ekson. Ekson Gen Gen affisert (HGNC (HGNC Transkript Ekson** Fenotype av symbol) ID) segdup* AAAS 13666 NM_015665.6 1-16 Achalasia-addisonianism-alacrimia syndrome, 231550 AARS 20 NM_001605.2 2-21 Epileptic encephalopathy, early infantile, 29 616339 AARS2 21022 NM_020745.4 1-22 Combined oxidative phosphorylation deficiency 8, 614096 AASS 17366 NM_005763.4 2-24 Hyperlysinaemia (Disorders of histidine, tryptophan or lysine metabolism) ABAT 23 NM_020686.6 2-16 GABA transaminase deficiency (Disorders of neurotransmitter metabolism, gamma-aminobutyrate)