Surface Characterization and Reactivity of Methylammionium Lead Iodide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bimetallic Catalyst Catalyzed Carbonylation of Methanol to Acetic Acid
materials Article Study on Rh(I)/Ru(III) Bimetallic Catalyst Catalyzed Carbonylation of Methanol to Acetic Acid Shasha Zhang 1, Wenxin Ji 1,2,*, Ning Feng 2, Liping Lan 1, Yuanyuan Li 1,2 and Yulong Ma 1,2 1 College of Chemistry and Chemical Engineering, Ningxia University, Yinchuan 750021, China; [email protected] (S.Z.); [email protected] (L.L.); [email protected] (Y.L.); [email protected] (Y.M.) 2 State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering, Ningxia University, Yinchuan 750021, China; [email protected] * Correspondence: [email protected]; Tel.: +86-135-1957-9989; Fax: +86-951-206-2323 Received: 13 July 2020; Accepted: 3 September 2020; Published: 11 September 2020 Abstract: In this study, a Rh(I)/Ru(III) catalyst with a bimetallic space structure was designed and synthesized. The interaction between the metals of the bimetallic catalyst and the structure of the bridged dimer can effectively reduce the steric hindrance effect and help speed up the reaction rate while ensuring the stability of the catalyst. X-ray photoelectron spectroscopy (XPS) results show that rhodium accepts electrons from chlorine, thereby increasing the electron-rich nature of rhodium and improving the catalytic activity. This promotes the nucleophilic reaction of the catalyst with methyl iodide and reduces the reaction energy barrier. The methanol carbonylation performance of the Rh/Ru catalyst was evaluated, and the results show that the conversion rate of methyl acetate and the yield of acetic acid are 96.0% under certain conditions. Furthermore, during the catalysis, no precipitate is formed and the amount of water is greatly reduced. -

Step-By-Step Guide to Better Laboratory Management Practices
Step-by-Step Guide to Better Laboratory Management Practices Prepared by The Washington State Department of Ecology Hazardous Waste and Toxics Reduction Program Publication No. 97- 431 Revised January 2003 Printed on recycled paper For additional copies of this document, contact: Department of Ecology Publications Distribution Center PO Box 47600 Olympia, WA 98504-7600 (360) 407-7472 or 1 (800) 633-7585 or contact your regional office: Department of Ecology’s Regional Offices (425) 649-7000 (509) 575-2490 (509) 329-3400 (360) 407-6300 The Department of Ecology is an equal opportunity agency and does not discriminate on the basis of race, creed, color, disability, age, religion, national origin, sex, marital status, disabled veteran’s status, Vietnam Era veteran’s status or sexual orientation. If you have special accommodation needs, or require this document in an alternate format, contact the Hazardous Waste and Toxics Reduction Program at (360)407-6700 (voice) or 711 or (800) 833-6388 (TTY). Table of Contents Introduction ....................................................................................................................................iii Section 1 Laboratory Hazardous Waste Management ...........................................................1 Designating Dangerous Waste................................................................................................1 Counting Wastes .......................................................................................................................8 Treatment by Generator...........................................................................................................12 -

Chapter -1 Introduction, Review of Literature And
CHAPTER -1 INTRODUCTION, REVIEW OF LITERATURE AND SCOPE OF THE PRESENT WORK 1 INTRODUCTION The heterocycles play an important part in the metabolism of all living cells and find important applications in industry.1 Among these important substances, such vitamins and coenzymes precursors as thiamine, riboflavin, nicotinic acid, adenine, biotin, vitamin Bi2, vitamin E, photosynthesizing pigment chlorophyll, the oxygen-transporting pigment haemoglobin, the purine and pyrimidine which are the components of the nucleic acids, and their metabolic products such as uric acid, allantoin, and alloxan to the amino-acids like histidine, tryptophan, proline, and the harmones such as kinetin, zeatin, heteroauxin and histamine contain heterocyclic ring system in them. Many of the drugs, natural as well as synthetic, which are in regular use are heterocyclic compounds. The several natural drugs such as alkaloids, the cardiac glycosides and antibiotics such as penicillin contain heterocyclic ring in them. Some other synthetic heterocyclic compounds are numerous and include barbiturates, thiouracil, carbimazole, 9-aminoacridine, 8-hydroxyquinoline, and vasoprassor modifiers. Polyvinylpyrrolidone are used as a replacement for serum lost in haemorrhage and shock. Many pesticides and weed killers such as paraquat, diquat and simazine; insecticides such as rotenone, diazinon, menazon; anthelminitics such as phenothiazine, thiabendazoles; rodenticides such as warfarin are heterocyclic compounds. The heterocycles are acting as the antidotes for poisoning due to the phosphorus insecticides, such as pyridine-2-aldoxime methiodide. The current use of the heterocycles in drugs and pesticides is due to the high resistance of heterocyclic substances to biological degradation. In addition to the drug value the synthetic heterocyclic compounds acts as chemotherapeutic agents, dyestuffs and co-polymers. -

Organic Chemistry – Ii
MANONMANIAM SUNDARANAR UNIVERSITY DIRECTORATE OF DISTANCE & CONTINUING EDUCATION, TIRUNELVELI III Year Major 1 – ORGANIC CHEMISTRY – II CONTENTS Unit I STEREOCHEMISTRY Unit II POLYNUCLEAR HYDROCARBONS Unit III HETEROCYCLIC COMPOUNDS Unit IV ALKALOIDS AND TERPENOIDS Unit V ORGANIC SPECTROSCOPY Page 1 MANONMANIAM SUNDARANAR UNIVERSITY DIRECTORATE OF DISTANCE & CONTINUING EDUCATION, TIRUNELVELI UNIT – I STEREOCHEMISTRY Stereoisomerism – definition – classification into optical and geometrical isomerism. Projection Formulae – Fischer, Sawhorse and Newman projection formulae – Notation of Optical isomers – D-L notation – Cahn – Ingold – Prelog rules – R-S notations for optical isomers. Optical isomerism – optical activity- optical and specific rotations – conditions for optical activity – asymmetric centre – chirality – achiral molecules – meaning of (+) and (-) Elements of symmetry – Racemisation – methods of recamisation. Resolution – methods of resolution (mechanical, seeding, biochemical and conversion to diastereoisomers). Optical activity in compounds not containing asymmetric carbon atoms.Biphenyls.allenes and spiranes. Geometrical isomerism – cis-trans, and E-Z notations – Geomertical isomerism in maleic and fumaric acids – Methods of distinguishing geometrical isomers using melting point, dipole moment, dehydration and cyclisation. UNIT – II POLYNUCLEAR HYDROCARBONS Isolatedsystems Preparation of dipheny1, dipheny1 methane, tri phenyl methane and stilbene. Condensed system Page 2 MANONMANIAM SUNDARANAR UNIVERSITY DIRECTORATE OF DISTANCE -

AMINE HYDROHALIDE EXTRACTION STUDIES by W
Wmm lili mS U t 2!582.e anni EUROPEAUJfJtLAiNN A1UJYLLLATOMIC. LLINJIJKATENERGYI ^UMJYLUINCOMMUNITl 1 Yï - EURATO.E,UJ\A1<^IVM1 ¡ty» *PiB'!«tflfc*'lni!" Γ· · if .'"TJ'f ΙΒ^',",·Ρ* i*""j(jM >'*>>ι·4· ΦΙΐΡτ mmé iiii «HRPS MINE HYDROHALIDE EXTRACTION &âW Transplutonium Elements Program lia Report UCRL No. 16254 Lawrence Radiation Laboratory - University of California Berkeley, Cal. - USA Euratom/Université de Liège Contract No. 003-61-2 TPUB AEC Contract No. W-7405-eng-48 Paper presented at the ir&mi Sì«»·» Gordon Research Conference on Ion Exchange -M New London, New Hampshire, USA - August 2-6, 1965 mm-, it:m. This document was prepared under the sponsorship of the Commission of the European Atomic Energy Community Neither the EURATOM Commission, its contractors nor any person acting on their behalf : Turi Make any warranty or representation, express or implied, with respect to the accuracy, completeness, or usefulness of the information, contained in this document, or that the use of any information, apparatus, method, or process disclosed in this document may not infringe privately owned rights ; or Assume any liability with respect to the use of, or for damages resulting from the use of any information, apparatus, method or process disclosed in this document. M·. EUR 2582.e AMINE HYDROHALIDE EXTRACTION STUDIES by W. MÜLLER (Euratom) European Atomic Energy Community - EURATOM Transplutonium Elements Program Report UCRL No. 16254, Lawrence Radiation Laboratory University of California, Berkeley, Cal. (USA) Euratom/Université de Liège Contract No. 003-61-2 TPUB AEC Contract No. W-7405-eng-48 Paper presented at the "Gordon Research Conference on Ion Exchange" New London, New Hampshire, USA - August 2-6, 1965 Brussels, December 1965 - 22 Pages - 10 Figures - FB 40 Equilibria between trilaurylamine dissolved in different organic diluents and aqueous dilute hydrohalic acids (HCl, HBr, HI) have been studied. -

Reduction of the Nitro Group to Amine by Hydroiodic Acid to Synthesize O-Aminophenol Derivatives As Putative Degradative Markers of Neuromelanin
Molecules 2014, 19, 8039-8050; doi:10.3390/molecules19068039 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Article Reduction of the Nitro Group to Amine by Hydroiodic Acid to Synthesize o-Aminophenol Derivatives as Putative Degradative Markers of Neuromelanin Kazumasa Wakamatsu 1,*, Hitomi Tanaka 1, Keisuke Tabuchi 1, Makoto Ojika 2, Fabio A. Zucca 3, Luigi Zecca 3 and Shosuke Ito 1 1 Department of Chemistry, Fujita Health University School of Health Sciences, Toyoake, Aichi 470-1192, Japan; E-Mails: [email protected] (H.T.); [email protected] (K.T.); [email protected] (S.I.) 2 Department of Applied Molecular Biosciences, Graduate School of Bioagricultural Sciences, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8601, Japan; E-Mail: [email protected] 3 Institute of Biomedical Technologies, National Research Council of Italy, Via Cervi, 93, Segrate, Milano 20090, Italy; E-Mails: [email protected] (F.A.Z.); [email protected] (L.Z.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +81-562-93-2518; Fax: +81-562-93-4595. Received: 4 May 2014; in revised form: 5 June 2014 / Accepted: 6 June 2014 / Published: 16 June 2014 Abstract: Neuromelanin (NM) is produced in dopaminergic neurons of the substantia nigra (SN) and in noradrenergic neurons of the locus coeruleus (LC). The synthesis of NM in those neurons is a component of brain aging and there is the evidence that this pigment can be involved in the pathogenesis of neurodegenerative diseases such as Parkinson’s disease. -

Chemical Compatibility Guide - Polyethylene for Ultratech Spill Containment Products
Chemical Compatibility Guide - Polyethylene For UltraTech Spill Containment Products This listing was prepared to provide guidance to the chemical compatibility When considering an UltraTech polyethylene product for use in secondary of UltraTech Spill Containment Products which are manufactured and containment applications, it is important to note that most secondary constructed of a molded polyethylene. containment products are designed to hold leaked chemicals for only hours, Polyethylene is susceptible to attack by some chemicals which may cause a day, at most a week. stress cracking, swelling, oxidation or may permeate the polyethylene. These secondary containment units would then be cleaned of any chemical. These reactions may reduce the physical properties of polyethylene. In these short term applications, a greater variety of chemicals may be used with the polyethylene since the exposure time of the chemical to the polyethylene is limited. A = Suitable for long term storage at 100 degrees Fahrenheit or less. B = Suitable for short term storage less than one year. C = Do NOT store these chemicals in UltraTech containment products. User testing may prove some of these chemicals are suitable for secondary containment applications with an exposure time of one week or less. Acetaldehyde (40%) ������������������������������������������A Ascetic Acid (50%) ���������������������������������������������A Carbon Bisulfide ������������������������������������������������C Acetamide ����������������������������������������������������������A -

Health Consultation
HEALTH CONSULTATION Potential Health Effects at a Clandestine Methamphetamine Laboratory using the Red Phosphorus Production Method Harrison, Clare County, MICHIGAN Prepared by Michigan Department of Community Health Under a Cooperative Agreement with Agency for Toxic Substances and Disease Registry ABBREVIATIONS AND ACRONYMS ACGIH American Conference of Governmental Industrial Hygienists AEGL Acute Exposure Guideline Level ATSDR Agency for Toxic Substances and Disease Registry BAYANET Bay Area Narcotics Enforcement Team cm2 centimeters squared (“square centimeters”) CMDHD Central Michigan District Health Department HCl hydrochloric acid (hydrogen chloride) HI hydroiodic acid (hydrogen iodide) IDLH Immediately Dangerous to Life and Health m3 meters cubed (“cubic meters”) MDCH Michigan Department of Community Health MDEQ Michigan Department of Environmental Quality meth methamphetamine, methamphetamine hydrochloride mg milligram MSP Michigan State Police NIOSH National Institute of Occupational Safety & Health OSHA Occupational Safety & Health Administration PEL permissible exposure level ppm parts per million RADS Reactive Airway Dysfunction Syndrome REL recommended exposure level TLV Threshold Limit Value µg microgram VOC volatile organic chemical (compound) i FOREWORD The federal Agency for Toxic Substances and Disease Registry (ATSDR) and the Michigan Department of Community Health (MDCH) have a cooperative agreement for conducting assessments and consultations regarding potential health hazards at toxic chemical contamination sites -

List of Chemical Resistance of Materials in Contact with Liquid ANALYSETTE 22 Next & ANALYSETTE 28 Imagesizer LASER PARTICLE SIZER & PARTICLE SIZER
List of chemical resistance of materials in contact with liquid ANALYSETTE 22 NeXT & ANALYSETTE 28 ImageSizer LASER PARTICLE SIZER & PARTICLE SIZER This chart information is based on FRITSCH's knowledge and experience and are intended as a guide and not as a guarantee. We cannot assure the perfect performance of the materials as that depends on many factors as working pressure, pressure picks, fluid, ambient temperature, fluid temperature, concentra‐ tion, duration of exposure, etc… Fritsch GmbH Milling and Sizing Industriestrasse 8 D - 55743 Idar-Oberstein Telephone: +49 (0) 6784/ 70-0 Email: [email protected] Internet: www.fritsch.de Version 04/2020 Index 001 Table of contents Table of contents 1 Materials in contact with liquid.................................................... 4 2 Abbreviations................................................................................. 5 3 List of chemical resistance............................................................. 7 3.1 KEY......................................................................................... 7 3.2 Note....................................................................................... 7 3.3 Table 1.................................................................................... 7 3.4 Table 2.................................................................................. 97 3.5 Table 3................................................................................ 105 - 3 - Materials in contact with liquid 1 Materials in contact with liquid Wet dispersion -

(10) Patent No.: US 6420304 B1
USOO6420304B1 (12) United States Patent (10) Patent No.: US 6,420,304 B1 Tsai et al. (45) Date of Patent: Jul. 16, 2002 (54) POLYMER-SUPPORTED CARBONYLATION 5,364.963 A 11/1994 Minami et al. ............. 502/326 CATALYST AND ITS USE 5,442,107 A * 8/1995 Beevor et al. 5,455.874 A * 10/1995 Ormsby et al. ............. 358/433 (75) Inventors: Chia-Jung Tsai, Yao-Lung Liu, both 5,466,874. A * 11/1995 Scates et al. of Kaohsiung, Hsi-Chin Tsai, Chiya-Yi, 6,066,762 A * 5/2000 Yoneda et al. - - - - - - - - - - - - - - 562/517 all of (TW) FOREIGN PATENT DOCUMENTS (73) Assignee: China Petrochemical Development EP O277824 * 10/1988 Corporation, Taipei (TW) * cited by examiner (*) Notice: Subject to any disclaimer, the term of this Primary Examiner Paul J. Killos patent is extended or adjusted under 35 ASSistant Examiner Robert W. Deemie U.S.C. 154(b) by 0 days. (74) Attorney, Agent, or Firm-Bacon & Thomas (57) ABSTRACT (21) Appl. No.: 09/553,054 The invention relates to a polymer-Supported carbonylation (22) Filed: Apr. 20, 2000 catalyst and its use in a process for preparing organic (51) Int. Cl."7 .......................... B01J 21/02; CO7C 51/14 carboxvlicy acid or anhvdridey having9. n+1 carbon atoms. The s CO7C 51/56s invention relates also to a pprocess for preparing Orgorganic carboxylic acid or anhydride having n+1 carbon atoms by (52) U.S. Cl. ....................... 502/207; 502/105; 502/151; carbonylating with monoxide, in the presence of the above 502/152; 502/159; 502/200; 502/313; 562/518; mentioned carbonylation catalyst, on alcohols having in 562/517; 562/497; 562/606; 562/607; 562/890; carbon atoms, ethers having 2n carbon atoms or esters 562/891; 562/519 formed from Said alcohols and acids. -

Chemical Formula Writing & Nomenclature Binary Acids Naming Binary Acids
© Mr. D. Scott; CHS Chemical Formula Writing & Nomenclature Binary Acids Ternary Acids Basic Rules for acids All formulas begin with hydrogen, H Those hydrogens listed first are the ones capable of being ionized when in solution and/or reacted with bases. Hydrogens listed later in the formula are not capable of being ionized. Binary Acids These follow the format HxAy where A represents some non-metal element covalently bonded to hydrogen. Naming Binary Acids hydro- -ic acid add prefix root of non-metal name This naming approach assumes that the acid compound in aqueous (dissolved in water). When these compounds are pure, the naming rules follow those of the binary molecules that we will cover later. Until further notice, we will name these compounds as acids in aqueous solution. © Mr. D. Scott; CHS The oxidation state of the non-metal follows the same rule as it does when it is combining with a metal. It will be the first one listed and negative. Examples: +1 -3 H + P H + P H3P hydrophosphoric acid +1 -2 H + S H + S H2S hydrosulfuric acid +1 -2 H + Se H + Se H2Se hydroselenic acid H + F H+1 + F-1 HF hydrofluoric acid H + Cl H+1 + Cl-1 HCl hydrochloric acid H + Br H+1 + Br-1 HBr hydrobromic acid H + I H+1 + I-1 HI hydroiodic acid © Mr. D. Scott; CHS Ternary Acids These follow the format Hx(polyatomic ion)y An important consideration – All acids are molecules. While we are using our PAL to combine various polyatomic ions with hydrogen, these are NOT IONIC COMPOUNDS. -
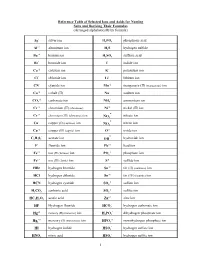
1 Reference Table of Selected Ions and Acids for Naming Salts and Deriving Their Formulas
Reference Table of Selected Ions and Acids for Naming Salts and Deriving Their Formulas (Arranged alphabetically by formula) + Ag silver ion HP34O phosphoric acid +3 Al aluminum ion HS2 hydrogen sulfide +2 Ba barium ion HS24O sulfuric acid Br- bromide ion I- iodide ion Ca+2 calcium ion K+ potassium ion Cl- chloride ion Li+ lithium ion CN- cyanide ion Mn+2 manganese (II) (manganous) ion Co+2 cobalt (II) Na+ sodium ion -2 + CO3 carbonate ion NH4 ammonium ion Cr+2 chromium (II) (chromous) Ni+2 nickel (II) ion - Cr+3 chromium (III) (chromous) ion nitrate ion NO3 - Cu+ copper (I) (cuprous) ion nitrite ion NO2 Cu+2 copper (II) (cupric) ion O-2 oxide ion - - C23H02 acetate ion OH hydroxide ion F- fluoride ion Pb+2 lead ion +2 -3 Fe iron (II) (ferrous) ion PO4 phosphate ion Fe+3 iron (III) (ferric) ion S-2 sulfide ion HBr hydrogen bromide Sn+2 tin (II) (stannous) ion HCl hydrogen chloride Sn+4 tin (IV) (stannic) ion -2 HCN hydrogen cyanide SO4 sulfate ion -2 HC23O carbonic acid SO3 sulfite ion +2 HC232HO acetic acid Zn zinc ion - HF Hydrogen fluoride HCO3 hydrogen carbonate ion +2 - Hg mercury (II) (mercuric) ion HP24O dihydrogen phosphate ion +2 -2 Hg2 mercury (I) (mercurous) ion HPO4 monohydrogen phosphate ion - HI hydrogen iodide HSO4 hydrogen sulfate ion - HNO3 nitric acid HSO3 hydrogen sulfite ion 1 NAMES AND FORMULAS FOR SOME COMMON ACIDS AND ANIONS (BASES) DERIVED FROM THEM HBr Hydrobromic acid Br- bromide ion (Hydrogen bromide) HCl Hydrochloric acid Cl- chloride ion (Hydrogen chloride) HCN Hydrocyanic acid CN - cyanide ion (Hydrogen cyanide)