Adenomatous Polyposis Coli Is Required for Early Events in the Normal Growth and Differentiation of the Developing Cerebral Cortex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Deregulated Wnt/Β-Catenin Program in High-Risk Neuroblastomas Without
Oncogene (2008) 27, 1478–1488 & 2008 Nature Publishing Group All rights reserved 0950-9232/08 $30.00 www.nature.com/onc ONCOGENOMICS Deregulated Wnt/b-catenin program in high-risk neuroblastomas without MYCN amplification X Liu1, P Mazanek1, V Dam1, Q Wang1, H Zhao2, R Guo2, J Jagannathan1, A Cnaan2, JM Maris1,3 and MD Hogarty1,3 1Division of Oncology, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA; 2Department of Biostatistics and Epidemiology, University of Pennsylvania School of Medicine, Philadelphia, PA, USA and 3Department of Pediatrics, University of Pennsylvania School of Medicine, Philadelphia, PA, USA Neuroblastoma (NB) is a frequently lethal tumor of Introduction childhood. MYCN amplification accounts for the aggres- sive phenotype in a subset while the majority have no Neuroblastoma (NB) is a childhood embryonal malig- consistently identified molecular aberration but frequently nancy arising in the peripheral sympathetic nervous express MYC at high levels. We hypothesized that acti- system. Half of all children with NB present with features vated Wnt/b-catenin (CTNNB1) signaling might account that define their tumorsashigh riskwith poor overall for this as MYC is a b-catenin transcriptional target and survival despite intensive therapy (Matthay et al., 1999). multiple embryonal and neural crest malignancies have A subset of these tumors are characterized by high-level oncogenic alterations in this pathway. NB cell lines without genomic amplification of the MYCN proto-oncogene MYCN amplification express higher levels of MYC and (Matthay et al., 1999) but the remainder have no b-catenin (with aberrant nuclear localization) than MYCN- consistently identified aberration to account for their amplified cell lines. -

Towards an Integrated View of Wnt Signaling in Development Renée Van Amerongen and Roel Nusse*
HYPOTHESIS 3205 Development 136, 3205-3214 (2009) doi:10.1242/dev.033910 Towards an integrated view of Wnt signaling in development Renée van Amerongen and Roel Nusse* Wnt signaling is crucial for embryonic development in all animal Notably, components at virtually every level of the Wnt signal species studied to date. The interaction between Wnt proteins transduction cascade have been shown to affect both β-catenin- and cell surface receptors can result in a variety of intracellular dependent and -independent responses, depending on the cellular responses. A key remaining question is how these specific context. As we discuss below, this holds true for the Wnt proteins responses take shape in the context of a complex, multicellular themselves, as well as for their receptors and some intracellular organism. Recent studies suggest that we have to revise some of messengers. Rather than concluding that these proteins are shared our most basic ideas about Wnt signal transduction. Rather than between pathways, we instead propose that it is the total net thinking about Wnt signaling in terms of distinct, linear, cellular balance of signals that ultimately determines the response of the signaling pathways, we propose a novel view that considers the receiving cell. In the context of an intact and developing integration of multiple, often simultaneous, inputs at the level organism, cells receive multiple, dynamic, often simultaneous and of both Wnt-receptor binding and the downstream, sometimes even conflicting inputs, all of which are integrated to intracellular response. elicit the appropriate cell behavior in response. As such, the different signaling pathways might thus be more intimately Introduction intertwined than previously envisioned. -

Hippocampus Formation: an Intriguing Collaboration Henk Roelink
bb10g06.qxd 04/03/2000 01:10 Page R279 Dispatch R279 Hippocampus formation: An intriguing collaboration Henk Roelink Recent genetic studies have shown that the signalling temporal lobes are formed. In an adult rodent, the hip- factor Wnt3a is required for formation of the pocampus is consequently still found close to the dorsal hippocampus; the developmental consequences of Wnt midline where it is initially formed (Figure 1). signalling in the hippocampus are mediated by multiple HMG-box transcription factors, with LEF-1 being It is clear that Wnt family members are required for the required just for formation of the dentate gyrus. development of many embryonic structures, functions mediated by their effects on fundamental processes such Address: Department of Biological Structure and Center for Developmental Biology, University of Washington, Box 357420, as cell proliferation, differentiation, survival or mainte- Seattle, Washington 98117-7420, USA. nance. Obtaining a clear picture of how each Wnt acts is E-mail: [email protected] complicated by the relatively large size of the family, which has at least 18 members in amniotes [3]. Analyses of Current Biology 2000, 10:R279–R281 mice lacking one or several Wnt genes have revealed some 0960-9822/00/$ – see front matter remarkable phenotypes, often characterized by failure of © 2000 Elsevier Science Ltd. All rights reserved. induction of very specific anatomical structures. Wnt3A is no exception, and although several papers have been pub- Inductive interactions are fundamental to the formation of lished already on the more obvious defects of Wnt3a all brain structures. and signalling by molecules of the knockout mice, a recent paper from McMahon and col- Wingless/Wnt family is known to play a role in many of leagues [1] has reported a very interesting defect in the them. -

A Uniform Human Wnt Expression Library Reveals a Shared Secretory Pathway and Unique Signaling Activities
Differentiation 84 (2012) 203–213 Contents lists available at SciVerse ScienceDirect Differentiation journal homepage: www.elsevier.com/locate/diff A uniform human Wnt expression library reveals a shared secretory pathway and unique signaling activities Rani Najdi a, Kyle Proffitt b, Stephanie Sprowl a, Simran Kaur b, Jia Yu b, Tracy M. Covey b, David M. Virshup b,1, Marian L. Waterman a,n a Department of Microbiology and Molecular Genetics, University of California, Irvine, CA, USA b Program in Cancer and Stem Cell Biology, Duke-NUS Graduate Medical School, Singapore article info abstract Article history: Wnt ligands are secreted morphogens that control multiple developmental processes during embry- Received 24 April 2012 ogenesis and adult homeostasis. A diverse set of receptors and signals have been linked to individual Accepted 16 June 2012 Wnts, but the lack of tools for comparative analysis has limited the ability to determine which of these Available online 9 July 2012 signals are general for the entire Wnt family, and which define subsets of differently acting ligands. We Keywords: have created a versatile Gateway library of clones for all 19 human Wnts. An analysis comparing Wnt signaling epitope-tagged and untagged versions of each ligand shows that despite their similar expression at the b-catenin mRNA level, Wnts exhibit considerable variation in stability, processing and secretion. At least 14 out of Wnt modification and secretion the 19 Wnts activate b-catenin-dependent signaling, an activity that is cell type-dependent and tracks with the stabilization of b-catenin and LRP6 phosphorylation. We find that the core Wnt modification and secretion proteins Porcupine (PORCN) and Wntless (WLS) are essential for all Wnts to signal through b-catenin-dependent and independent pathways. -

Wnt3a: Functions and Implications in Cancer Sha He1,2, Yi Lu1,2, Xia Liu1,2, Xin Huang1,2, Evan T
He et al. Chin J Cancer (2015) 34:50 DOI 10.1186/s40880-015-0052-4 REVIEW Open Access Wnt3a: functions and implications in cancer Sha He1,2, Yi Lu1,2, Xia Liu1,2, Xin Huang1,2, Evan T. Keller3, Chao‑Nan Qian4* and Jian Zhang1,2,3* Abstract Wnt3a, one of Wnt family members, plays key roles in regulating pleiotropic cellular functions, including self-renewal, proliferation, differentiation, and motility. Accumulating evidence has suggested that Wnt3a promotes or suppresses tumor progression via the canonical Wnt signaling pathway depending on cancer type. In addition, the roles of Wnt3a signaling can be inhibited by multiple proteins or chemicals. Herein, we summarize the latest findings on Wnt3a as an important therapeutic target in cancer. Keywords: Wnt3a, The Wnt signaling pathway, Cancer Background: the Wnt signaling pathway cell polarity pathway (Wnt-PCP pathway) and the Wnt- The Wnt gene was first identified in 1982 and is named calcium pathway (Wnt-Ca2+ pathway) [5]. According to the Int gene in mice [1]. A subsequent study reported the characteristics of its functions, the Wnt family can that the Int gene and wingless gene in Drosophila were also be divided into two categories: Wnt1 and Wnt5a. homologous genes [2]. Therefore, both genes were rec- The Wnt1 category includes Wnt1, Wnt2, Wnt2b, Wnt3, ognized as the Wnt gene [3]. The Wnt family comprises Wnt3a, Wnt7a, Wnt8, Wnt8b, and Wnt10a, which are 19 human proteins, including Wnt1, Wnt2, Wnt2b involved in the canonical signaling pathway, whereas (Wnt13), Wnt3, Wnt3a, Wnt4, Wnt5a, Wnt5b, Wnt6, Wnt4, Wnt5a, and Wnt11 belong to the Wnt5a category Wnt7a, Wnt7b, Wnt8a, Wnt8b, Wnt9a (Wnt14), Wnt9b and activate the noncanonical signaling pathway. -

Analysis of the Canonical WNT Pathway Simona Giunta Department of Biological Sciences, Brunel University, Uxbridge, Middlesex, UK
05-giunta 25-02-2010 15:21 Pagina 187 ACTA BIOMED 2009; 80: 187-199 © Mattioli 1885 R EVIEW A gust of WNT: analysis of the canonical WNT pathway Simona Giunta Department of Biological Sciences, Brunel University, Uxbridge, Middlesex, UK Abstract. The Wnt pathway is a signal-transduction cascade that mediates communication between cells; the Wnt pathway is involved in key steps during embryological development and in the maintenance of adult tissue homeostasis. Mutational dysregulation of Wnt cascade components has been observed in diverse hu- man pathological conditions and in oncogenic transformations. For these reasons, the Wnt signalling path- way has acquired growing interest in scientific and medical research over recent years. This review outlines the biochemical and functional features of the Wnt cascade with particular emphasis on a detailed function- al analysis of all key players. In this instance, the regulations of the pathway have also been covered, empha- sizing novelty in this regard. Furthermore, past and present studies on Wnt have been included, as well as a prediction of scientific progress, which may be made in this rapidly evolving field, in the near future; the re- view also embraces considerations on how further understanding of the Wnt pathway will provide important insight into managing human diseases. (www.actabiomedica.it) Key words: Wnt canonical pathway, b-catenin, signal transduction, haematopoietic stem cells, carcinogenesis Introduction molecules which regulate the main steps of embryoge- nesis. Multicellular organisms necessitate a communi- The Wnt family of signalling proteins has been cation system to grow and function; in complex mul- found to be involved in embryonic patterning, in the ticellular organisms, like humans, cell-to-cell commu- homeostasis of adult tissue self-renewal and in the nication becomes the basis of life. -

WNT8B Antibody (C-Term) Blocking Peptide Synthetic Peptide Catalog # Bp13948b
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 WNT8B Antibody (C-term) Blocking peptide Synthetic peptide Catalog # BP13948b Specification WNT8B Antibody (C-term) Blocking WNT8B Antibody (C-term) Blocking peptide - peptide - Background Product Information The WNT gene family consists of structurally Primary Accession Q93098 related geneswhich encode secreted signaling proteins. These proteins have beenimplicated in oncogenesis and in several developmental WNT8B Antibody (C-term) Blocking peptide - Additional Information processes,including regulation of cell fate and patterning duringembryogenesis. This gene is a member of the WNT gene family. Itencodes a Gene ID 7479 protein which shows 95%, 86% and 71% amino acid identityto the mouse, zebrafish and Other Names Xenopus Wnt8B proteins, respectively.The Protein Wnt-8b, WNT8B expression patterns of the human and mouse genes appearidentical and are restricted to the Target/Specificity developing brain. Thechromosomal location of The synthetic peptide sequence used to generate the antibody AP13948b was this gene to 10q24 suggests it as acandidate selected from the C-term region of WNT8B. gene for partial epilepsy. A 10 to 100 fold molar excess to antibody is recommended. Precise conditions should be WNT8B Antibody (C-term) Blocking optimized for a particular assay. peptide - References Format Katoh, M., et al. Int. J. Oncol. Peptides are lyophilized in a solid powder 30(5):1273-1277(2007)Grupe, A., et al. Am. J. format. Peptides can be reconstituted in Hum. Genet. 78(1):78-88(2006)Deloukas, P., et solution using the appropriate buffer as al. Nature 429(6990):375-381(2004)Saitoh, T., needed. -

Wnt1 and Wnt10b Function Redundantly at the Zebrafish Midbrain
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Available online at www.sciencedirect.com R Developmental Biology 254 (2003) 172–187 www.elsevier.com/locate/ydbio wnt1 and wnt10b function redundantly at the zebrafish midbrain–hindbrain boundary Arne C. Lekven,a,* Gerri R. Buckles,a Nicholas Kostakis,b and Randall T. Moonb a Department of Biology, Texas A&M University, 3258 TAMU, College Station, TX 77843-3258, USA b Howard Hughes Medical Institute, Department of Pharmacology and Center for Developmental Biology, Box 357750, University of Washington School of Medicine, Seattle, WA 98195, USA Received for publication 28 May 2002, revised 8 October 2002, accepted 1 November 2002 Abstract Wnt signals have been shown to be involved in multiple steps of vertebrate neural patterning, yet the relative contributions of individual Wnts to the process of brain regionalization is poorly understood. Wnt1 has been shown in the mouse to be required for the formation of the midbrain and the anterior hindbrain, but this function of wnt1 has not been explored in other model systems. Further, wnt1 is part of a Wnt cluster conserved in all vertebrates comprising wnt1 and wnt10b, yet the function of wnt10b during embryogenesis has not been explored. Here, we report that in zebrafish wnt10b is expressed in a pattern overlapping extensively with that of wnt1. We have generated a deficiency allele for these closely linked loci and performed morpholino antisense oligo knockdown to show that wnt1 and wnt10b provide partially redundant functions in the formation of the midbrain–hindbrain boundary (MHB). -
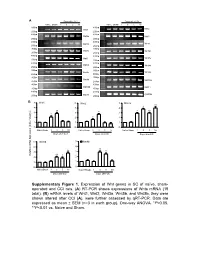
Supplementary Figure 1. Expression of Wnt Genes in SC of Naïve
A Days after CCI Days after CCI Naive Sham 1 3 5 10 Naive Sham 1 3 5 10 500bp 400bp Wnt1 Wnt2 250bp 250bp 400bp 400bp Wnt2b Wnt3 250bp 250bp 400bp 400bp Wnt3a Wnt4 250bp 250bp 300bp 400bp Wnt5a Wnt5b 200bp 250bp 400bp 400bp Wnt6 Wnt7a 250bp 250bp 400bp 400bp Wnt7b Wnt8a 250bp 250bp 400bp 350bp Wnt8b Wnt9a 250bp 200bp 400bp 400bp Wnt9b Wnt10a 250bp 250bp 400bp 400bp Wnt10b Wnt11 250bp 250bp 400bp 400bp Wnt16 GAPDH 250bp 250bp B 5 Wnt1 5 Wnt2 5 Wnt3a ** 4 4 4 * ** ** 3 3 * 3 * * 2 2 * 2 1 1 1 0 0 0 NaïveSham 1 3 5 10 Naïve Sham 1 3 5 10 Naïve Sham 1 3 5 10 Days after CCI Days after CCI Days after CCI 5 Wnt5b 5 Wnt8b 4 4 ** 3 ** 3 ** Relative mRNA Expression (fold of change) ** 2 * 2 1 1 0 0 NaïveSham 1 3 5 10 NaïveSham 1 3 5 10 Days after CCI Days after CCI Supplementary Figure 1 A B Negative Weak Moderate Strong Negative Weak Moderate Strong Large-sized cells 100% 100% 80% 80% 60% 60% 40% 20% 40% 0% 20% Sham CCI-1d CCI-14d Medium-sized cells Wnt3a immunoreactivity cells 0% 100% CGRP(+) IB4(+) 80% Small cells 60% 40% 20% 0% Sham CCI-1d CCI-14d Wnt3a immunoreactivity cells Small cells 100% 80% 60% 40% 20% 0% Sham CCI-1d CCI-14d Fig. S2 4 Fz1 4 Fz3 4 Fz4 4 Fz5 ** 3 3 ** 3 3 2 ** 2 2 ** 2 * 1 1 1 1 0 0 0 0 NaïveSham 1 5 10 NaïveSham 1 5 10 Naïve Sham 1 5 10 NaïveSham 1 5 10 Days after CCI Days after CCI Days after CCI Days after CCI 4 Fz6 4 Fz7 4 Fz8 4 Fz9 (Fold of Change) of (Fold 3 3 3 ** 3 Relative mRNA Expression Expression mRNA Relative ** 2 2 2 ** 2 * 1 1 1 1 0 0 0 0 NaïveSham 1 5 10 NaïveSham 1 5 10 Naïve Sham 1d 5d 10d NaïveSham 1 -

Oncogenic Wnt3a
Journal of Cancer 2019, Vol. 10 5862 Ivyspring International Publisher Journal of Cancer 2019; 10(23): 5862-5873. doi: 10.7150/jca.31599 Research Paper Oncogenic Wnt3a: A Candidate Specific Marker and Novel Molecular Target for Hepatocellular Carcinoma Wenjie Zheng1*, Min Yao2*, Miao Fang1, Liuhong Pan1, Li Wang3, Junling Yang1, Zhizhen Dong4, Dengfu Yao1 1. Research Center of Clinical Medicine, Affiliated Hospital of Nantong University, Nantong 226001, Jiangsu Province, China; 2. Department of Medical Immunology, Medical School of Nantong University, Nantong 226001, Jiangsu Province, China; 3. Department of Medical Informatics, Medical School of Nantong University, Nantong 226001, Jiangsu Province, China; 4. Department of Diagnostics, Affiliated Hospital of Nantong University, Nantong 226001, Jiangsu Province, China. * Equal contributors Corresponding author: Prof. Dengfu Yao, M.D. & Ph.D., Research Center of Clinical Medicine, Affiliated Hospital of Nantong University, 20 West Temple Rd, Jiangsu 226001, China. Tel: +0086-513-85052523; Fax: +0086-513-85052254; E-mail: [email protected] © The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions. Received: 2019.06.12; Accepted: 2019.08.20; Published: 2019.10.08 Abstract Background and aim: It is of the utmost importance for the specific diagnosis and effective therapy of hepatocellular carcinoma (HCC). Abnormality of oncogenic Wingless-type MMTV integration site family member 3a (Wnt3a) has been associated with progression of HCC. In this study, we aimed to evaluate Wnt3a as a novel biomarker and target for HCC. Methods: Circulating Wnt3a levels were quantitatively detected in a cohort of chronic liver diseases by an enzyme-linked immune-absorbent assay (ELISA). -

Wnt Signaling Pathway Pharmacogenetics in Non-Small Cell Lung Cancer
The Pharmacogenomics Journal (2014) 14, 509–522 & 2014 Macmillan Publishers Limited All rights reserved 1470-269X/14 www.nature.com/tpj ORIGINAL ARTICLE Wnt signaling pathway pharmacogenetics in non-small cell lung cancer DJ Stewart1, DW Chang2,YYe2, M Spitz2,CLu3, X Shu2, JA Wampfler4, RS Marks5, YI Garces6, P Yang4 and X Wu2 Wingless-type protein (Wnt)/b-catenin pathway alterations in non-small cell lung cancer (NSCLC) are associated with poor prognosis and resistance. In 598 stage III–IV NSCLC patients receiving platinum-based chemotherapy at the MD Anderson Cancer Center (MDACC), we correlated survival with 441 host single-nucleotide polymorphisms (SNPs) in 50 Wnt pathway genes. We then assessed the most significant SNPs in 240 Mayo Clinic patients receiving platinum-based chemotherapy for advanced NSCLC, 127 MDACC patients receiving platinum-based adjuvant chemotherapy and 340 early stage MDACC patients undergoing surgery alone (cohorts 2–4). In multivariate analysis, survival correlates with SNPs for AXIN2 (rs11868547 and rs4541111, of which rs11868547 was assessed in cohorts 2–4), Wnt-5B (rs12819505), CXXC4 (rs4413407) and WIF-1 (rs10878232). Median survival was 19.7, 15.6 and 10.7 months for patients with 1, 2 and 3–5 unfavorable genotypes, respectively (P ¼ 3.8 Â 10 À 9). Survival tree analysis classified patients into two groups (median survival time 11.3 vs 17.3 months, P ¼ 4.7 Â 10 À 8). None of the SNPs achieved significance in cohorts 2–4; however, there was a trend in the same direction as cohort 1 for 3 of the SNPs. Using online databases, we found rs10878232 displayed expression quantitative trait loci correlation with the expression of LEMD3, a neighboring gene previously associated with NSCLC survival. -

Neuroretina Specification in Mouse Embryos Requires Six3- Mediated Suppression of Wnt8b in the Anterior Neural Plate
Neuroretina specification in mouse embryos requires Six3- mediated suppression of Wnt8b in the anterior neural plate Wei Liu, … , Milan Jamrich, Guillermo Oliver J Clin Invest. 2010;120(10):3568-3577. https://doi.org/10.1172/JCI43219. Research Article Development Retinal degeneration causes vision impairment and blindness in humans. If one day we are to harness the potential of stem cell–based cell replacement therapies to treat these conditions, it is imperative that we better understand normal retina development. Currently, the genes and mechanisms that regulate the specification of the neuroretina during vertebrate eye development remain unknown. Here, we identify sine oculis–related homeobox 3 (Six3) as a crucial player in this process in mice. In Six3 conditional–mutant mouse embryos, specification of the neuroretina was abrogated, but that of the retinal pigmented epithelium was normal. Conditional deletion of Six3 did not affect the initial development of the optic vesicle but did arrest subsequent neuroretina specification. Ectopic rostral expansion of Wnt8b expression was the major response to Six3 deletion and the leading cause for the specific lack of neuroretina, as ectopicW nt8b expression in transgenic embryos was sufficient to suppress neuroretina specification. Using chromatin immunoprecipitation assays, we identified Six3-responsive elements in the Wnt8b locus and demonstrated that Six3 directly repressed Wnt8b expression in vivo. Our findings provide a molecular framework to the program leading to neuroretina differentiation and may be relevant for the development of novel strategies aimed at characterizing and eventually treating different abnormalities in eye formation. Find the latest version: https://jci.me/43219/pdf Research article Neuroretina specification in mouse embryos requires Six3-mediated suppression of Wnt8b in the anterior neural plate Wei Liu,1 Oleg Lagutin,1 Eric Swindell,2 Milan Jamrich,3 and Guillermo Oliver1 1Department of Genetics and Tumor Cell Biology, St.