Soil, Plant and Water Reference Methods for the Western Region1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sloping and Benching Systems
Trenching and Excavation Operations SLOPING AND BENCHING SYSTEMS OBJECTIVES Upon the completion of this section, the participant should be able to: 1. Describe the difference between maximum allowable slope and actual slope. 2. Observe how the angle of various sloped systems varies with soil type. 3. Evaluate layered systems to determine the proper trench slope. 4. Illustrate how shield systems and sloping systems interface in combination systems. ©HMTRI 2000 Page 42 Trenching REV1 Trenching and Excavation Operations SLOPING SYSTEMS If enough surface room is available, sloping or benching the trench walls will offer excellent protection without any additional equipment. Cutting the slope of the excavation back to its prescribed angle will allow the forces of cohesion (if present) and internal friction to hold the soil together and keep it from flowing downs the face of the trench. The soil type primarily determines the excavation angle. Sloping a method of protecting employees from caveins by excavating to form sides of an excavation that are inclined away from the excavations so as to prevent caveins. In practice, it may be difficult to accurately determine these sloping angles. Most of the time, the depth of the trench is known or can easily be determined. Based on the vertical depth, the amount of cutback on each side of the trench can be calculated. A formula to calculate these cutback distances will be included with each slope diagram. NOTE: Remember, the beginning of the cutback distance begins at the toe of the slope, not the center of the trench. Accordingly, the cutback distance will be the same regardless of how wide the trench is at the bottom. -
SECTION A-A NTS 2" Grade Board Notches
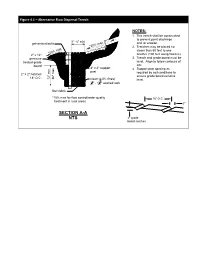
Figure 4.1 – Alternative Flow Dispersal Trench NOTES: 1. This trench shall be constructed to prevent point discharge 1' - 6" min galvanized bolts and /or erosion. *20% max 2. Trenches may be placed no closer than 50 feet to one *20% max 2" x 12" another (100 feet along flowline). pressure 3. Trench and grade board must be treated grade level. Align to follow contours of board site. 4" x 4" support 4. Support post spacing as post required by soil conditions to 2" x 2" notches ensure grade board remains 18" O.C. 36" max 12" min. clean (< 5% fines) level. 3 1 4" - 1 2" washed rock filter fabric *15% max for flow control/water quality 18" O.C. treatment in rural areas 2" SECTION A-A NTS 2" grade board notches Figure 4.2 – Pipe Compaction Design and Backfill O.D. O.D. limits of pipe W (see note 4) 3' max. 3' max. compaction limit of pipe zone 1' - 0" bedding material for flexible 1' - 0" pipe (see 0.15 O.D. min. note 6) B O.D. limits of pipe compaction 0.65 O.D. min. foundation level A* gravel backfill gravel backfill for for pipe bedding foundations when specified * A = 4" min., 27" I.D. and under 6" min., over 27" I.D. A. Metal and Concrete Pipe Bedding for Flexible Pipe span span span 3' max Flexible Pipe NOTES: 3' max 3' max 1. Provide uniform support under barrels. 1'-0" 2. Hand tamp under haunches. 3. Compact bedding material to 95% max. -

Simple Soil Tests for On-Site Evaluation of Soil Health in Orchards
sustainability Article Simple Soil Tests for On-Site Evaluation of Soil Health in Orchards Esther O. Thomsen 1, Jennifer R. Reeve 1,*, Catherine M. Culumber 2, Diane G. Alston 3, Robert Newhall 1 and Grant Cardon 1 1 Dept. Plant Soils and Climate, Utah State University, Logan, UT 84322, USA; [email protected] (E.O.T.); [email protected] (R.N.); [email protected] (G.C.) 2 UC Cooperative Extension, Fresno, CA 93710, USA; [email protected] 3 Dept. Biology, Utah State University, Logan, UT 84322, USA; [email protected] * Correspondence: [email protected] Received: 20 September 2019; Accepted: 26 October 2019; Published: 29 October 2019 Abstract: Standard commercial soil tests typically quantify nitrogen, phosphorus, potassium, pH, and salinity. These factors alone are not sufficient to predict the long-term effects of management on soil health. The goal of this study was to assess the effectiveness and use of simple physical, biological, and chemical soil health indicator tests that can be completed on-site. Analyses were conducted on soil samples collected from three experimental peach orchards located on the Utah State Horticultural Research Farm in Kaysville, Utah. All simple tests were correlated to comparable lab analyses using Pearson’s correlation. The highest positive correlations were found between Solvita®respiration, and microbial biomass (R = 0.88), followed by our modified slake test and microbial biomass (R = 0.83). Both Berlese funnel and pit count methods of estimating soil macro-organism diversity were fairly predictive of soil health. Overall, simple commercially available chemical tests were weak indicators of soil nutrient concentrations compared to laboratory tests. -

Frequency and Magnitude of Selected Historical Landslide Events in The
Chapter 9 Frequency and Magnitude of Selected Historical Landslide Events in the Southern Appalachian Highlands of North Carolina and Virginia: Relationships to Rainfall, Geological and Ecohydrological Controls, and Effects Richard M. Wooten , Anne C. Witt , Chelcy F. Miniat , Tristram C. Hales , and Jennifer L. Aldred Abstract Landsliding is a recurring process in the southern Appalachian Highlands (SAH) region of the Central Hardwood Region. Debris fl ows, dominant among landslide processes in the SAH, are triggered when rainfall increases pore-water pressures in steep, soil-mantled slopes. Storms that trigger hundreds of debris fl ows occur about every 9 years and those that generate thousands occur about every 25 years. Rainfall from cyclonic storms triggered hundreds to thousands of debris R. M. Wooten (*) Geohazards and Engineering Geology , North Carolina Geological Survey , 2090 US Highway 70 , Swannanoa , NC 28778 , USA e-mail: [email protected] A. C. Witt Virginia Department of Mines, Minerals and Energy , Division of Geology and Mineral Resources , 900 Natural Resources Drive, Suite 500 , Charlottesville , VA 22903 , USA e-mail: [email protected] C. F. Miniat Coweeta Hydrologic Lab , Center for Forest Watershed Research, USDA Forest Service, Southern Research Station , 3160 Coweeta Lab Road , Otto , NC 28763 , USA e-mail: [email protected] T. C. Hales Hillslope Geomorphology , School of Earth and Ocean Sciences, Cardiff University , Main Building, Park Place , Cardiff CF10 3AT , UK e-mail: [email protected] J. L. Aldred Department of Geography and Earth Sciences , University of North Carolina at Charlotte , 9201 University City Blvd. , Charlotte , NC 28223 , USA e-mail: [email protected] © Springer International Publishing Switzerland 2016 203 C.H. -

Soil Testing Can Help Nutrient Deficiencies Or Imbalances
Do You Have Problems With: • Nutrient deficiencies in crops • Poor plant growth and response from applied fertilizers • Hard to manage weeds • Low crop yields • Poor quality forages • Irregular plant growth in your fields • Managing manure or compost applications Soil tests help to identify production problems related to Soil Testing Can Help nutrient deficiencies or imbalances. Above: Nitrogen defi- ciency in corn (photo: Ryan Stoffregen, Illinois). Below: Phosphorus deficiency in corn. Source: www.ipni.net Benefits of Soil Testing: • Determines nutrient levels in the soil • Determines pH levels (lime needs) • Provides a decision making tool to determine what nutrients to apply and how much • Potential for higher yielding crops • Potential for higher quality crops • More efficient fertilizer use Costs: Generally soil tests cost $7 to $10.00 per sample. The costs of soil tests vary depending on: 1. Your state (some states offer free soil testing) 2. The lab that is used. 3. The items being tested for (the cost increases as more nutrients are being analyzed). NOTE: Some state agencies and land grant universities provide free soil testing for the basic soil test items (pH, available phosphorus, potassium, calcium, and magnesium, and organic matter). Additional costs may be charged for testing for micronutrients. In other states, all soil testing is done by private labs and generally charge $7-$10 for the basic test. One soil test should be taken for each field, or for each 20 acres within a field. See example on page 3. Soil Testing How Often Should I Soil Test? Generally, you should soil test every 3-5 years or more often if manure is applied or you are trying to make large nutrient or pH changes in the soil. -

Division 2 Earthwork
Division 2 Earthwork 2-01 Clearing, Grubbing, and Roadside Cleanup 2-01.1 Description The Contractor shall clear, grub, and clean up those areas staked or described in the Special Provisions. This Work includes protecting from harm all trees, bushes, shrubs, or other objects selected to remain. “Clearing” means removing and disposing of all unwanted material from the surface, such as trees, brush, down timber, or other natural material. “Grubbing” means removing and disposing of all unwanted vegetative matter from underground, such as sod, stumps, roots, buried logs, or other debris. “Roadside cleanup”, whether inside or outside the staked area, means Work done to give the roadside an attractive, finished appearance. “Debris” means all unusable natural material produced by clearing, grubbing, or roadside cleanup. 2-01.2 Disposal of Usable Material and Debris The Contractor shall meet all requirements of state, county, and municipal regulations regarding health, safety, and public welfare in the disposal of all usable material and debris. The Contractor shall dispose of all debris by one or more of the disposal methods described below. 2-01.2(1) Disposal Method No. 1 – Open Burning The open burning of residue resulting from land clearing is restricted by Chapter 173-425 of the Washington Administrative Code (WAC). No commercial open burning shall be conducted without authorization from the Washington State Department of Ecology or the appropriate local air pollution control authority. All burning operations shall be strictly in accordance with these authorizations. 2-01.2(2) Disposal Method No. 2 – Waste Site Debris shall be hauled to a waste site obtained and provided by the Contractor in accordance with Section 2-03.3(7)C. -

Soil Test Handbook for Georgia
SOIL TEST HANDBOOK FOR GEORGIA Georgia Cooperative Extension College of Agricultural & Environmental Sciences The University of Georgia Athens, Georgia 30602-9105 EDITORS: David E. Kissel Director, Agricultural and Environmental Services Laboratories & Leticia Sonon Program Coordinator, Soil, Plant, & Water Laboratory The University of Georgia and Ft. Valley State University, the U.S. Department of Agriculture and counties of the state cooperating. The Cooperative Extension, the University of Georgia College of Agricultural and Environmental Sciences offers educational programs, assistance and materials to all people without regard to race, color, national origin, age, sex or disability. An Equal Opportunity Employer/Affirmative Action Organization Committed to a Diverse Work Force Issued in furtherance of Cooperative Extension work, Acts of May 8 and June 30, 1914, The University of Georgia College of Agricultural and Environmental Sciences and the U.S. Department of Agriculture cooperating. Dr. Scott Angle, Dean and Director Special Bulletin 62 September 2008 i TABLE OF CONTENTS INTRODUCTION .......................................................................................................................................................2 SOIL TESTING...........................................................................................................................................................4 SOIL SAMPLING .......................................................................................................................................................4 -

3 Groundwork and Foundations
P1: JZW/JZZ BY032-03 BY032-Emmitt BY032-v1.cls September 13, 2004 9:6 3 Groundwork and Foundations The foundation of a building is that part of walls, piers and columns in direct contact with, and transmitting loads to, the ground. The building founda- tion is sometimes referred to as the artificial foundation, and the ground on which it bears as the natural foundation. Early buildings were founded on rock or firm ground; it was not until the beginning of the twentieth century that concrete was increasingly used as a foundation base for walls. With the introduction of local and then national building regulations, standard forms of concrete foundations have become accepted practice in the UK, along with more rigorous investigation of the nature and bearing capacity of soils and bedrock. 3.1 Functional requirements The primary functional requirement of a foundation is strength and stability. Strength and stability The combined, dead, imposed and wind loads on a building must be transmit- ted to the ground safely, without causing deflection or deformation of the build- ing or movement of the ground that would impair the stability of the building and/or neighbouring structures. Foundations should also be designed and constructed to resist any movements of the subsoil. Foundations should be designed so that any settlement is both limited and uniform under the whole of the building. Some settlement of a building on a soil foundation is inevitable. As the building is erected the loads placed on the foundation increase and the soil is compressed. This settlement should be limited to avoid damage to service pipes and drains connected to the building. -
Trench Blasting with DYNAMITE a TRADITION of INNOVATION
Trench Blasting with DYNAMITE A TRADITION OF INNOVATION Dyno Nobel’s roots reach back to every significant in- novation in explosives safety and technology. Today, Dyno Nobel supplies a full line of explosives products and blasting services to mines, quarries and contractors in nearly every part of the world. DYNAMITE PRODUCT OF CHOICE FOR TRENCH BLASTING One explosive product has survived the test of time to become a true classic in the industry. DYNAMITE! The dynamite products manufactured today by Dyno Nobel are similar to Alfred Nobel’s original 1860s invention yet, in selected applications, they outperform any other commercial explosives on the market. The high energy, reliability and easy loading characteristics of dynamite make it the product of choice for difficult and demand- ing trench blasting jobs. Look to Unigel®, Dynomax Pro® and Unimax® to make trench blasting as effective and efficient as it can be. DISCLAIMER The information set forth herein is provided for informational purposes only. No representation or warranty is made or intended by DYNO NOBEL INC. or its affiliates as to the applicability of any procedures to any par- ticular situation or circumstance or as to the completeness or accuracy of any information contained herein. User assumes sole responsibility for all results and consequences. ® Cover photo depicts a trench blast using Primacord detonating cord, MS ® Connectors and Unimax dynamite. SAFE BLASTING REMINDERS Blasting safety is our first priority. Review these remind- ers frequently and make safety your first priority, too. • Dynamite products will provide higher energy value than alternate products used for trenching due to their superior energy, velocity and weight strength. -

Fact Sheet: Trenching and Excavation Safety
Fact sheet Trenching and excavation safety Between 2016 and 2019, three Minnesota workers died in a trench, excavation or ground collapse. Cave-ins pose the greatest risk and are much more likely than other excavation-related accidents to result in worker fatalities. Minnesota law requires employers to provide a workplace free of recognized hazards that may cause serious injury or death. This includes the trenching and excavation requirements of 29 CFR 1926.651 and 1926.652. An excavation is any man-made cut, cavity, trench or depression in an earth surface, formed by earth removal. A trench – or a trench excavation – is a narrow excavation (in relation to its length) made below the surface of the ground; in general, the depth is greater than the width, but the width of a trench (measured at the bottom) is not greater than 15 feet. Dangers of trenching and excavation In addition to cave-ins, other potential hazards include falls, falling loads, hazardous atmospheres and incidents involving mobile equipment. Two cubic yards of soil can weigh about 6,000 pounds or as much as a small car. An unprotected trench is an early grave. Do not enter an unprotected trench. Trench safety measures Trenches five feet deep or greater require a protective system unless the excavation is made entirely in stable rock. If the trench is fewer than five feet deep, a competent person may determine a protective system is not required. Trenches 20 feet deep or greater require that the protective system be designed by a registered professional engineer or be based on tabulated data prepared and/or approved by a registered professional engineer in accordance with 1926.652(b) and (c). -

Addressing Pasture Compaction
Addressing Pasture Compaction Weighing the Pros and Cons of Two Options UVM Project team: Dr. Josef Gorres, Dr. Rachel Gilker, Jennifer Colby, Bridgett Jamison Hilshey Partners: Mark Krawczyk (Keyline Vermont) and farmers Brent & Regina Beidler, Guy & Beth Choiniere, John & Rocio Clark, Lyle & Kitty Edwards, and Julie Wolcott & Stephen McCausland Writing: Josef Gorres, Rachel Gilker and Jennifer Colby • Design and Layout: Jennifer Colby • Photos: Jennifer Colby and Rachel Gilker Additional editing by Cheryl Herrick and Bridgett Jamison Hilshey Th is is dedicated to our farmer partners; may our work help you farm more productively, profi tably, and ecologically. VT Natural Resources Conservation Service UVM Center for Sustainable Agriculture http://www.vt.nrcs.usda.gov http://www.uvm.edu/sustainableagriculture UVM Plant & Soil Science Department http://pss.uvm.edu Financial support for this project and publication was provided through a VT Natural Resources Conservation Service (NRCS) Conservation Innovation Grant (CIG). We thank VT-NRCS for their eff orts to build a 2 strong natural resource foundation for Vermont’s farming systems. Introduction A few years ago, some grass-based dairy farmers came to us with the question, “You know, what we really need is a way to fi x the compaction in pastures.” We started digging for answers. Th is simple request has led us on a lively journey. We began by adapting methods to alleviate compac- tion in other climates and cropping systems. We worked with fi ve Vermont dairy farmers to apply these practices to their pastures, where other farmers could come and observe them in action. We assessed the pros and cons of these approaches and we are sharing those results and observations here. -

Slope Stability
Slope stability Causes of instability Mechanics of slopes Analysis of translational slip Analysis of rotational slip Site investigation Remedial measures Soil or rock masses with sloping surfaces, either natural or constructed, are subject to forces associated with gravity and seepage which cause instability. Resistance to failure is derived mainly from a combination of slope geometry and the shear strength of the soil or rock itself. The different types of instability can be characterised by spatial considerations, particle size and speed of movement. One of the simplest methods of classification is that proposed by Varnes in 1978: I. Falls II. Topples III. Slides rotational and translational IV. Lateral spreads V. Flows in Bedrock and in Soils VI. Complex Falls In which the mass in motion travels most of the distance through the air. Falls include: free fall, movement by leaps and bounds, and rolling of fragments of bedrock or soil. Topples Toppling occurs as movement due to forces that cause an over-turning moment about a pivot point below the centre of gravity of the unit. If unchecked it will result in a fall or slide. The potential for toppling can be identified using the graphical construction on a stereonet. The stereonet allows the spatial distribution of discontinuities to be presented alongside the slope surface. On a stereoplot toppling is indicated by a concentration of poles "in front" of the slope's great circle and within ± 30º of the direction of true dip. Lateral Spreads Lateral spreads are disturbed lateral extension movements in a fractured mass. Two subgroups are identified: A.