Developing New Afm Imaging Technique and Software for Dna Mismatch Repair
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Design and Implementation of Image Processing and Compression Algorithms for a Miniature Embedded Eye Tracking System Pavel Morozkin
Design and implementation of image processing and compression algorithms for a miniature embedded eye tracking system Pavel Morozkin To cite this version: Pavel Morozkin. Design and implementation of image processing and compression algorithms for a miniature embedded eye tracking system. Signal and Image processing. Sorbonne Université, 2018. English. NNT : 2018SORUS435. tel-02953072 HAL Id: tel-02953072 https://tel.archives-ouvertes.fr/tel-02953072 Submitted on 29 Sep 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Sorbonne Université Institut supérieur d’électronique de Paris (ISEP) École doctorale : « Informatique, télécommunications & électronique de Paris » Design and Implementation of Image Processing and Compression Algorithms for a Miniature Embedded Eye Tracking System Par Pavel MOROZKIN Thèse de doctorat de Traitement du signal et de l’image Présentée et soutenue publiquement le 29 juin 2018 Devant le jury composé de : M. Ales PROCHAZKA Rapporteur M. François-Xavier COUDOUX Rapporteur M. Basarab MATEI Examinateur M. Habib MEHREZ Examinateur Mme Maria TROCAN Directrice de thèse M. Marc Winoc SWYNGHEDAUW Encadrant Abstract Human-Machine Interaction (HMI) progressively becomes a part of coming future. Being an example of HMI, embedded eye tracking systems allow user to interact with objects placed in a known environment by using natural eye movements. -

Pipenightdreams Osgcal-Doc Mumudvb Mpg123-Alsa Tbb
pipenightdreams osgcal-doc mumudvb mpg123-alsa tbb-examples libgammu4-dbg gcc-4.1-doc snort-rules-default davical cutmp3 libevolution5.0-cil aspell-am python-gobject-doc openoffice.org-l10n-mn libc6-xen xserver-xorg trophy-data t38modem pioneers-console libnb-platform10-java libgtkglext1-ruby libboost-wave1.39-dev drgenius bfbtester libchromexvmcpro1 isdnutils-xtools ubuntuone-client openoffice.org2-math openoffice.org-l10n-lt lsb-cxx-ia32 kdeartwork-emoticons-kde4 wmpuzzle trafshow python-plplot lx-gdb link-monitor-applet libscm-dev liblog-agent-logger-perl libccrtp-doc libclass-throwable-perl kde-i18n-csb jack-jconv hamradio-menus coinor-libvol-doc msx-emulator bitbake nabi language-pack-gnome-zh libpaperg popularity-contest xracer-tools xfont-nexus opendrim-lmp-baseserver libvorbisfile-ruby liblinebreak-doc libgfcui-2.0-0c2a-dbg libblacs-mpi-dev dict-freedict-spa-eng blender-ogrexml aspell-da x11-apps openoffice.org-l10n-lv openoffice.org-l10n-nl pnmtopng libodbcinstq1 libhsqldb-java-doc libmono-addins-gui0.2-cil sg3-utils linux-backports-modules-alsa-2.6.31-19-generic yorick-yeti-gsl python-pymssql plasma-widget-cpuload mcpp gpsim-lcd cl-csv libhtml-clean-perl asterisk-dbg apt-dater-dbg libgnome-mag1-dev language-pack-gnome-yo python-crypto svn-autoreleasedeb sugar-terminal-activity mii-diag maria-doc libplexus-component-api-java-doc libhugs-hgl-bundled libchipcard-libgwenhywfar47-plugins libghc6-random-dev freefem3d ezmlm cakephp-scripts aspell-ar ara-byte not+sparc openoffice.org-l10n-nn linux-backports-modules-karmic-generic-pae -

Nanosurf Operating Instructions (Naio).Book
Nanosurf NaioAFM Operating Instructions for Naio Control Software Version 3.6 “NANOSURF” AND THE NANOSURF LOGO ARE TRADEMARKS OF NANOSURF AG, REGISTERED AND/OR OTHERWISE PROTECTED IN VARIOUS COUNTRIES. COPYRIGHT © NOVEMBER 2015, NANOSURF AG, SWITZERLAND. OPERATING INSTRUCTIONS V3.6R0, BT05386-22. Table of contents Table of contents PART A: INTRODUCTION TO THE INSTRUMENT CHAPTER 1: The NaioAFM 15 1.1: Introduction.............................................................................................. 16 1.2: Components of the system...................................................................... 16 1.2.1: Contents of the tool set ..................................................................................17 1.3: Connectors, indicators and controls....................................................... 18 CHAPTER 2: Installing the NaioAFM 21 2.1: Installing the Naio control software ....................................................... 22 2.1.1: Preparation..........................................................................................................22 2.1.2: Installation...........................................................................................................22 2.2: Installing the hardware............................................................................ 23 CHAPTER 3: Preparing for measurement 25 3.1: Introduction.............................................................................................. 26 3.2: Initializing the NaioAFM system ........................................................... -

A Robust Multilayer Image Steganography Method Based on a Randomized Lsb Algorithm
International Journal of Mechanical Engineering and Technology (IJMET) Volume 9, Issue 9, September 2018, pp. 272–284, Article ID: IJMET_09_09_031 Available online at https://iaeme.com/Home/issue/IJMET?Volume=9&Issue=9 ISSN Print: 0976-6340 and ISSN Online: 0976-6359 © IAEME Publication Scopus Indexed A ROBUST MULTILAYER IMAGE STEGANOGRAPHY METHOD BASED ON A RANDOMIZED LSB ALGORITHM Saad Almutairi Faculty of Computers and Information Technology, University of Tabuk, Tabuk City, Saudia Arabi Abdulgader Almutairi College of Sciences and arts in ArRass, Qassim University, Qassim, Kingdom of Saudi Arabia ABSTRACT Recently, the demand for the uses of the steganography techniques to exchange a hidden secret data throughout unsecure networks has increased substantially. Nonetheless, although there are many techniques and methods out there in steganography field, a further researches need to be conducted in order to come up with a new secure techniques and methods. Thus, our research proposes a new robust multilayer image steganography method called MITEGO method that is based on a randomized LSB algorithm to overcome the weaknesses of the previous related works in the field, and to provide more secure image steganography at different points. MITEGO method follows a set of rules to create a stego image object (stego file) to embed and extract a secret message based on two layers: Hardening Layer and Embed/Extract Layer. Subsequently, MiTEGOsoft software tool is developed based on MITEGO method. Finally, MITEGO method is evaluated based on experiments and analysis of corresponding results. The research calculated Peak Signal-to-Noise Ratio (PSNR) in decibel (dB) unit by using ImageMagick software tool for each generated stego file. -

Metodi Per L'analisi Di Immagini
Metodi per l’analisi di immagini AFM Prof. Claudio Rivetti Dipartimento di Biochimica e Biologia Molecolare Università degli Studi di Parma Corso di perfezionamento in TECNICHE DI MICROSCOPIA A FORZA ATOMICA - Parma 29 febbraio 2012 Sommario Che cosa è un’immagine e come viene rappresentata Tipi di immagine: RGB, Indexed, Grayscale, Black and white Formato dei file di immagini Filtri e trasformazioni per elaborare immagini AFM Riconoscimento e misura di oggetti Software disponibili Che cosa è un’immagine R G B R 79 R 141 R 112 G 46 G 109 G 78 B 16 B 21 B 20 R 112 R 248 R 241 G 80 G 216 G 184 B 48 B 104 B 104 R 56 R 207 R 241 G 24 G 116 G 148 B 8 B 60 B 48 2563 = 16.777.216 colori 256 colori Color Table Index R G B i 240 i 220 i 215 1 125 43 55 2 230 56 90 3 22 220 102 4 34 130 179 5 55 133 140 i 80 i 201 i 221 6 140 21 77 7 150 28 61 8 140 10 14 … … … … 254 8 89 98 i 4 i 208 i 180 255 220 100 199 256 11 202 251 I 54 I 114 I 85 I 87 I 219 I 195 I 33 I 141 I 172 0 1 0 0 11 0 1 0 Profondità del colore dell’immagine La profondità di colore (color depth) è la quantità di bit necessari per rappresentare il colore di un singolo pixel in un'immagine 1 bit 4 bit 1 bit 21 = 2 colori 2 bit 22 = 4 colori 4 bit 24 = 16 colori 8 bit 28 = 256 colori 12 bit 212 = 4096 colori 8 bit 24 bit 16 bit 216 = 65536 colori 24 bit 224 = 16.777.216 colori B G R I Un’immagine grayscale può essere resa a colori utilizzando una scala di falsi colori Salvare un immagine su disco in un file Contiene informazioni relative Header all’immagine Contiene la matrice o le matrici (nel caso di RGB) di numeri che rappresentano l’immagine Data La dimensione del file dipende, dalla dimensione dell’immagine, dal tipo di immagine (RGB, grayscale, indexed) dai byte utilizzati per rappresentare il valore dei pixel. -

Laserinduced Nanobubbles - Interaction of Gold Nanoparticles with Pulsed Laserlight”
Electrical Scanning Probe Microscopy on Organic Optoelectronic Structures Dissertation zur Erlangung des Grades Doktor der Naturwissenschaften\ " am Fachbereich Physik, Mathematik und Informatik der Johannes-Gutenberg-Universit¨at in Mainz vorgelegt von Stefan Weber geboren in Trier Mainz, den 26. August 2010 Die vorliegende Arbeit wurde in der Zeit von September 2007 bis August 2010 unter der Betreuung von Dr. Rudiger¨ Berger und Prof. Dr. Hans-Jurgen¨ Butt am Max- Planck-Institut fur¨ Polymerforschung in Mainz durchgefuhrt.¨ Tag der mundlichen¨ Prufung:¨ 3. November 2010 Dekan: Prof. Dr. Manfred Lehn 1. Berichterstatter: Prof. Dr. H.J. Butt 2. Berichterstatter: Prof. Dr. T. Palberg 3. Berichterstatter: Prof. Dr. E. Meyer (Universit¨at Basel, Schweiz) 4. Berichterstatter: Prof. Dr. D.Y. Yoon (Seoul National University, Sudkorea)¨ D77 \I am a firm believer that without speculation there is no good and original observation." Charles R. Darwin in a letter to Alfred R. Wallace, December 22nd, 1857 Zusammenfassung Im Bereich der organischen Optoelektronik hat die mikro- und nanoskopische Struk- tur der Materialien einen großen Einfluss auf die Leistungsf¨ahigkeit der Bauteile. In diesem Bereich gewinnen rasterkraftmikroskopische Methoden (SFM) immer mehr an Bedeutung. Neben topographischer Information k¨onnen zahlreiche andere Ober- fl¨acheneigenschaften wie elektrische Leitf¨ahigkeit (Leitf¨ahigkeits-Rasterkraftmikros- kopie, C-SFM) oder Oberfl¨achenpotentiale (Kelvinsondenmikroskopie, KPFM) mit Aufl¨osungen im Bereich von Nanometern gemessen werden. Im Rahmen dieser Arbeit wurden die SFM basierten elektrischen Betriebsmodi ver- wendet um den Zusammenhang zwischen Morphologie und den elektrischen Eigen- schaften in optoelektronischen Hybridstrukturen zu erforschen. Diese Strukturen wurden in der Gruppe von Prof. J. Gutmann (MPI-P Mainz) entwickelt. -
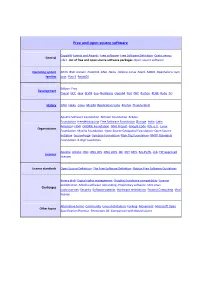
Free and Open Source Software
Free and open source software Copyleft ·Events and Awards ·Free software ·Free Software Definition ·Gratis versus General Libre ·List of free and open source software packages ·Open-source software Operating system AROS ·BSD ·Darwin ·FreeDOS ·GNU ·Haiku ·Inferno ·Linux ·Mach ·MINIX ·OpenSolaris ·Sym families bian ·Plan 9 ·ReactOS Eclipse ·Free Development Pascal ·GCC ·Java ·LLVM ·Lua ·NetBeans ·Open64 ·Perl ·PHP ·Python ·ROSE ·Ruby ·Tcl History GNU ·Haiku ·Linux ·Mozilla (Application Suite ·Firefox ·Thunderbird ) Apache Software Foundation ·Blender Foundation ·Eclipse Foundation ·freedesktop.org ·Free Software Foundation (Europe ·India ·Latin America ) ·FSMI ·GNOME Foundation ·GNU Project ·Google Code ·KDE e.V. ·Linux Organizations Foundation ·Mozilla Foundation ·Open Source Geospatial Foundation ·Open Source Initiative ·SourceForge ·Symbian Foundation ·Xiph.Org Foundation ·XMPP Standards Foundation ·X.Org Foundation Apache ·Artistic ·BSD ·GNU GPL ·GNU LGPL ·ISC ·MIT ·MPL ·Ms-PL/RL ·zlib ·FSF approved Licences licenses License standards Open Source Definition ·The Free Software Definition ·Debian Free Software Guidelines Binary blob ·Digital rights management ·Graphics hardware compatibility ·License proliferation ·Mozilla software rebranding ·Proprietary software ·SCO-Linux Challenges controversies ·Security ·Software patents ·Hardware restrictions ·Trusted Computing ·Viral license Alternative terms ·Community ·Linux distribution ·Forking ·Movement ·Microsoft Open Other topics Specification Promise ·Revolution OS ·Comparison with closed -
SPM Simulator Tutorial
走査プローブ顕微鏡シミュレータ Scanning Probe Microscope Simulator チュートリアルTutorial ver.β January 27, 2017 Advanced Algorithm & Systems Co., Ltd. Tohoku University, WPI-AIMR Contents Introduction ..................................................................................................................... 5 Composition of SPM simulator ........................................................................................ 6 Chapter 1 Installation of the software and preparation ........................ 11 1 installation of the software ............................................................................. 11 Chapter 2 Operation of the software .................................................... 20 1 Overview of the GUI ........................................................................................ 20 2 Part names and functions ................................................................................ 21 3 Start up GUI ..................................................................................................... 23 4 Operations of a project file .............................................................................. 24 Create a new project ............................................................................................................... 24 Load an existing project file .................................................................................................... 25 Show a project file ................................................................................................................. -

Gwyddion-User-Guide-En-2009-11-11
Gwyddion user guide Petr Klapetek, David Necas,ˇ and Christopher Anderson Version 2009-11-11. This guide is a work in progress. ii Gwyddion user guide Copyright © 2004–2007, 2009 Petr Klapetek, David Necas,ˇ Christopher Anderson Permission is granted to copy, distribute and/or modify this document under the terms of either • The GNU Free Documentation License, Version 1.2 or any later version published by the Free Software Foundation; with no Invariant Sections, no Front-Cover Texts, and no Back-Cover Texts. A copy of the license is included in the section entitled GNU Free Documentation License. • The GNU General Public License, Version 2 or any later version published by the Free Software Foundation. A copy of the license is included in the section entitled GNU General Public License. Contents iii Contents 1 Introduction 1 1.1 Motivation . .1 1.2 Licensing . .1 2 Installation 2 2.1 Linux/Unix Packages . .2 2.2 MS Windows Packages . .2 2.3 Build Dependencies . .3 2.4 Linux/Unix from Source Code Tarball . .4 Quick Instructions . .4 Source Unpacking . .4 Configuration . .4 Configuration tweaks . .5 Compilation . .5 Installation . .5 Deinstallation . .6 RPM Packages . .6 2.5 MacOSX.......................................................6 Preparation . .6 MacPorts . .6 Fink..........................................................7 Running . .7 2.6 MS Windows from Source Code Tarball . .7 Unpacking . .7 Configuration . .8 Compilation . .8 Installation . .8 Executable Installers . .9 2.7 Subversion Checkout, Development . .9 Linux/Unix . .9 MSWindows ..................................................... 10 3 Getting Started 11 3.1 Main Window . 11 3.2 Data Browser . 12 Controlling the Browser . 12 Channels . 13 Graphs......................................................... 13 Spectra . -

Gwyddion-User-Guide-En.Pdf
Gwyddion user guide Petr Klapetek, David Necas,ˇ and Christopher Anderson This guide is a work in progress. ii Gwyddion user guide Copyright © 2004–2007, 2009–2021 Petr Klapetek, David Necas,ˇ Christopher Anderson Permission is granted to copy, distribute and/or modify this document under the terms of either • The GNU Free Documentation License, Version 1.2 or any later version published by the Free Software Foundation; with no Invariant Sections, no Front-Cover Texts, and no Back-Cover Texts. A copy of the license is included in the section entitled GNU Free Documentation License. • The GNU General Public License, Version 2 or any later version published by the Free Software Foundation. A copy of the license is included in the section entitled GNU General Public License. Contents iii Contents 1 Introduction 1 1.1 Motivation . .1 1.2 Licensing . .1 2 Installation 2 2.1 Linux/Unix Packages . .2 2.2 MS Windows Packages . .2 Uninstallation . .2 Registry keys . .3 Missing features . .3 Enabling pygwy . .3 3 Getting Started 4 3.1 Main Window . .4 3.2 Data Browser . .5 Controlling the Browser . .6 Images.........................................................6 Graphs.........................................................6 Spectra . .6 Volume ........................................................6 XYZ..........................................................6 3.3 Managing Files . .6 File Loading . .7 File Merging . .8 File Saving . .8 Document History . .8 3.4 Data Window . .9 3.5 Graph Window . .9 3.6 Tools.......................................................... 10 3.7 False Color Mapping . 11 Color Range Tool . 12 Color Gradient Editor . 13 3.8 Presentations and Masks . 13 Presentations . 13 Masks......................................................... 14 3.9 Selections . 14 Selection Manager . 15 3.10 OpenGL 3D Data Display . -

AFM Characterization of Thin Films
Czech Metrology Institute, Czech Republic Use of Gwyddion libraries for your little tasks Petr Klapetek, David Nečas Anna Charvátová Campbell, Miroslav Valtr Gwyddion Open source software for SPM data analysis Gwyddion is Free and Open Source software for SPM data analysis working on GNU/Linux, Microsoft Windows, Mac OS X and FreeBSD operating systems on common architectures. It aims to provide multiplatform modular program for 2D data analysis that could be easily extended by modules and plug-ins. Development started in 2003, originally motivated by lack of sufficiently transparent data processing software for metrology purposes. After ten years it includes support for more that 100 file formats and all the typically used processing routines for SPM data processing (plus many not so typical algorithms as well). Basic Gwyddion structure Gwyddion relies on the GLib utility library for portability and uses GLib object system GObject for its own objects. Graphical user interface is implemented with the Gtk+ toolkit, with a fair amount of Gwyddion specific extension widgets. Gwyddion can be divided into four main components: 1. libraries, providing basic and advanced data processing routines, graphical user interface elements and other utility functions and objects, 2. the application, quite small and simple, serving primarily as a glue connecting the other components together in a common graphical interface, 3. modules, technically run-time loaded libraries, that provide most of the actual functionality and present it to the user, they often extensively use library methods, 4. plug-ins, standalone programs that are more independent of Gwyddion than modules, both technically and legally. It should be noted that plug-ins in their current form were found to be a bad idea and while they are still supported it is for compatibility. -

End-User License Agreement – V9.0
END-USER LICENSE AGREEMENT – V9.0 ACCEPTANCE Please read this document carefully. Before installing this product, you must accept the terms of this End-User License Agreement (hereinafter “EULA”). By installing or using the software or by clicking on “I Accept”, you (on behalf of yourself and the business entity you represent) agree that you have read all of the terms and conditions of this EULA and that you fully accept to be bound by these terms and conditions. This EULA applies to the then current version of the Product. DEFINITIONS Distributor: the commercial entity that has provided the Product to the End User. The Distributor may be a commercial distributor, but also directly the Editor and more frequently an instrument manufacturer that integrates the Product into their own offering. Documentation: All operating guides, and other reference materials, including in particular the Reference Manual, the Reference Guide, examples and tutorials. Dongle: small hardware device that must be plugged into a USB port on the PC in order to make it possible to run the Product. Network dongles are available for multiple licenses. Alternatively, the dongle can also be an equivalent piece of software. Editor: the developer and editor of the Product is the company DIGITAL SURF SARL potentially including its subsidiaries, whose registered office is 16 rue Lavoisier, 25000 Besançon, FRANCE. Effective Date: Date of the first launch of the product. End User: Individual user(s) employed by the company or organization that bought the Product from the Distributor. Fixed-term License: License to use the Product with a fixed expiry date, with a License term commencing on the Effective Date and ending as of the expiry of its subscription period.