Provides a Process of Hydrogenating 3-Hydroxypropionalde
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aldrich FT-IR Collection Edition I Library
Aldrich FT-IR Collection Edition I Library Library Listing – 10,505 spectra This library is the original FT-IR spectral collection from Aldrich. It includes a wide variety of pure chemical compounds found in the Aldrich Handbook of Fine Chemicals. The Aldrich Collection of FT-IR Spectra Edition I library contains spectra of 10,505 pure compounds and is a subset of the Aldrich Collection of FT-IR Spectra Edition II library. All spectra were acquired by Sigma-Aldrich Co. and were processed by Thermo Fisher Scientific. Eight smaller Aldrich Material Specific Sub-Libraries are also available. Aldrich FT-IR Collection Edition I Index Compound Name Index Compound Name 3515 ((1R)-(ENDO,ANTI))-(+)-3- 928 (+)-LIMONENE OXIDE, 97%, BROMOCAMPHOR-8- SULFONIC MIXTURE OF CIS AND TRANS ACID, AMMONIUM SALT 209 (+)-LONGIFOLENE, 98+% 1708 ((1R)-ENDO)-(+)-3- 2283 (+)-MURAMIC ACID HYDRATE, BROMOCAMPHOR, 98% 98% 3516 ((1S)-(ENDO,ANTI))-(-)-3- 2966 (+)-N,N'- BROMOCAMPHOR-8- SULFONIC DIALLYLTARTARDIAMIDE, 99+% ACID, AMMONIUM SALT 2976 (+)-N-ACETYLMURAMIC ACID, 644 ((1S)-ENDO)-(-)-BORNEOL, 99% 97% 9587 (+)-11ALPHA-HYDROXY-17ALPHA- 965 (+)-NOE-LACTOL DIMER, 99+% METHYLTESTOSTERONE 5127 (+)-P-BROMOTETRAMISOLE 9590 (+)-11ALPHA- OXALATE, 99% HYDROXYPROGESTERONE, 95% 661 (+)-P-MENTH-1-EN-9-OL, 97%, 9588 (+)-17-METHYLTESTOSTERONE, MIXTURE OF ISOMERS 99% 730 (+)-PERSEITOL 8681 (+)-2'-DEOXYURIDINE, 99+% 7913 (+)-PILOCARPINE 7591 (+)-2,3-O-ISOPROPYLIDENE-2,3- HYDROCHLORIDE, 99% DIHYDROXY- 1,4- 5844 (+)-RUTIN HYDRATE, 95% BIS(DIPHENYLPHOSPHINO)BUT 9571 (+)-STIGMASTANOL -

Chemical Trends in Solid Alkali Pertechnetates † ‡ ‡ ‡ ‡ ‡ Jamie Weaver, , Chuck Z
This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. Article pubs.acs.org/IC Chemical Trends in Solid Alkali Pertechnetates † ‡ ‡ ‡ ‡ ‡ Jamie Weaver, , Chuck Z. Soderquist, Nancy M. Washton, Andrew S. Lipton, Paul L. Gassman, § ∥ † † ‡ ⊥ Wayne W. Lukens, Albert A. Kruger, Nathalie A. Wall, and John S. McCloy*, , , † Department of Chemistry, Washington State University, Pullman, Washington 99164, United States ‡ Pacific Northwest National Laboratory, Richland, Washington 99352, United States § Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States ∥ U.S. Department of Energy (DOE), Office of River Protection, Richland, Washington 99352, United States ⊥ Materials Science and Engineering Program and School of Mechanical & Materials Engineering, Washington State University, Pullman, Washington 99164, United States *S Supporting Information ABSTRACT: Insight into the solid-state chemistry of pure technetium-99 (99Tc) oxides is required in the development of a robust immobilization and disposal system for nuclear waste stemming from the radiopharmaceutical industry, from the production of nuclear weapons, and from spent nuclear fuel. However, because of its radiotoxicity and the subsequent requirement of special facilities and handling procedures for research, only a few studies have been completed, many of which are over 20 years old. In this study, we report the synthesis of pure alkali pertechnetates (sodium, potassium, rubidium, and cesium) and analysis of these compounds by Raman spectroscopy, X-ray absorption spectroscopy (XANES and EXAFS), solid-state nuclear magnetic resonance (static and magic angle spinning), and neutron diffraction. The structures and spectral signatures of these compounds will aid in refining the understanding of 99Tc incorporation into and release from nuclear waste glasses. -

Harmonized Tariff Schedule of the United States (2020) Revision 14 Annotated for Statistical Reporting Purposes
Harmonized Tariff Schedule of the United States (2020) Revision 14 Annotated for Statistical Reporting Purposes SECTION VI PRODUCTS OF THE CHEMICAL OR ALLIED INDUSTRIES VI-1 Notes 1. (a) Goods (other than radioactive ores) answering to a description in heading 2844 or 2845 are to be classified in those headings and in no other heading of the tariff schedule. (b) Subject to paragraph (a) above, goods answering to a description in heading 2843, 2846 or 2852 are to be classified in those headings and in no other heading of this section. 2. Subject to note 1 above, goods classifiable in heading 3004, 3005, 3006, 3212, 3303, 3304, 3305, 3306, 3307, 3506, 3707 or 3808 by reason of being put up in measured doses or for retail sale are to be classified in those headings and in no other heading of the tariff schedule. 3. Goods put up in sets consisting of two or more separate constituents, some or all of which fall in this section and are intended to be mixed together to obtain a product of section VI or VII, are to be classified in the heading appropriate to that product, provided that the constituents are: (a) Having regard to the manner in which they are put up, clearly identifiable as being intended to be used together without first being repacked; (b) Entered together; and (c) Identifiable, whether by their nature or by the relative proportions in which they are present, as being complementary one to another. Additional U.S. Notes 1. In determining the amount of duty applicable to a solution of a single compound in water subject to duty in this section at a specific rate, an allowance in weight or volume, as the case may be, shall be made for the water in excess of any water of crystallization which may be present in the undissolved compound. -

P'aturan Harga Satuan Standar BATAN-2009
BADAN TENAGA NUKLIR NASIONAL PERATURAN KEPALA BADAN TENAGA NUKLIR NASIONAL NOMOR: 110/KA/VI/2008 VIII/2007 TENTANG HARGA SATUAN STANDAR BATAN TAHUN ANGGARAN 2009 DENGAN RAHMAT TUHAN YANG MAHA ESA KEPALA BADAN TENAGA NUKLIR NASIONAL, Menimbang : a. bahwa dengan Peraturan Kepala BATAN Nomor 122/KA/VIII/2007 telah ditetapkan Harga Satuan Standar BATAN Tahun Anggaran 2008; b. bahwa mengingat banyaknya perubahan jumlah maupun struktur biaya yang terjadi, akan berdampak terhadap pelaksanaan kegiatan Tahun Anggaran 2009; c. bahwa berdasarkan pertimbangan sebagaimana dimaksud dalam huruf a dan huruf b, perlu menetapkan Peraturan Kepala BATAN tentang Harga Satuan Standar BATAN Tahun Anggaran 2009; Mengingat : 1. Undang-Undang Nomor 17 Tahun 2003 tentang Keuangan Negara (Lembaran Negara Republik Indonesia Tahun 2003 Nomor 47, Tambahan Lembaran Negara Republik Indonesia Nomor 4286); 2. Undang-Undang Nomor 1 Tahun 2004 tentang Perbendaharaan Negara (Lembaran Negara Republik Indonesia Tahun 2004 Nomor 5, Tambahan Lembaran Negara Republik Indonesia Nomor 4355); 3. Undang-Undang Nomor 25 Tahun 2004 tentang Sistem Perencanaan Pembangunan Nasional (Lembaran Negara Republik Indonesia Tahun 2004 Nomor 104, Tambahan Lembaran Negara Republik Indonesia Nomor 4421); 4. Peraturan Pemerintah Nomor 21 Tahun 2004 tentang Penyusunan Rencana Kerja dan Anggaran Kementerian Negara/Lembaga (Lembaran Negara Republik Indonesia Tahun 2004 Nomor 75, Tambahan Lembaran Negara Republik Indonesia Nomor 4406); BADAN TENAGA NUKLIR NASIONAL -2- 5. Peraturan Pemerintah Nomor 77 Tahun 2005 tentang Jenis dan Tarif Atas Jenis Penerimaan Negara Bukan Pajak Yang Berlaku Pada Badan Tenaga Nuklir Nasional (Lembaran Negara Republik Indonesia Tahun 2005 Nomor 163, Tambahan Lembaran Negara Republik Indonesia Nomor 4591); 6. Keputusan Presiden Nomor 103 Tahun 2001 tentang Kedudukan, Tugas, Fungsi, Kewenangan, Susunan Organisasi, dan Tata Kerja Lembaga Pemerintah Non Departemen sebagaimana telah beberapa kali diubah, terakhir dengan Peraturan Presiden Nomor 64 Tahun 2005; 7. -

Circular of the Bureau of Standards No. 539 Volume 9: Standard X-Ray
jnal Bureau o Library, %■ ■ KARS i860 NBS CIRCULAR 539 VOLUME 9 ri / Standard X-ray Diffraction Powder Patterns UNITED STATES DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS THE NATIONAL BUREAU OF STANDARDS Functions and Activities The functions of the National Bureau of Standards are set forth in the Act of Congress, March 3, 1901, as amended by Congress in Public Law 619, 1950. These include the development and maintenance of the national standards of measurement and the provision of means and methods for making measurements consistent with these standards; the determination of physical constants and properties of materials; the development of methods and instruments for testing materials, devices, and structures; advisory services to government agencies on scientific and technical problems; in¬ vention and development of devices to serve special needs of the Government; and the development of standard practices, codes, and specifications. The work includes basic and applied research, development, engineering, instrumentation, testing, evaluation, calibration services, and various consultation and information services. Research projects are also performed for other government agencies when the work relates to and supplements the basic program of the Bureau or when the Bureau’s unique competence is required. The scope of activities is suggested by the listing of divi¬ sions and sections on the inside of the back cover. Publications The results of the Bureau’s work take the form of either actual equipment and devices or pub¬ lished -

ID Ammonium Perrhenate
ID Card Ammonium perrhenate Version 26 May 2014 Notes: • This ID card is used to support the substance sameness discussions in SIEFs and to describe the substance to the best of the SIEF members’ knowledge. • It also aims at grouping communications relevant to the request of available data or information, the approval of the proposed Lead Registrant and the registration strategy with the SIEF. • It is the responsibility of each individual registrant to identify their substance and to report company-specific identity in their Registration Dossier (section 1 of IUCLID). DISCLAIMER All data and information contained in this document shall be treated by the receiving party (i) in full confidence with the adequate respect of any confidential and/or proprietary nature of such information and (ii) only in the framework of the purpose of agreeing on substance sameness, Lead Registrant and overall REACH Strategy for the concerned Substance under REACH (the ‘Purpose’). The receiving party (and any representative) shall not be allowed to use or circulate any or all parts of this document for any other purpose than the Purpose, without the prior written consent of the European Precious Metals Federation (EPMF). The content provided in this document is given for the Purpose and as such, no guarantee or warranty whatsoever (expressed or implied) is given as to its accuracy, completeness, merchantability or fitness for any particular purpose which the receiving party may have. In any case, any use by the receiving party would be made at its sole risk and liability. 1. Identification of the substance Table 1. Identification of the substance Original (in EC inventory) Name Ammonium perrhenate EC number 237-075-6 CAS number 13598-65-7 Description Not available Composition type Mono-constituent substance European Precious Metals Federation aisbl p1 E: [email protected] W: www.epmf.be p1 ID Card 2. -

Staging Category Base Rate Article Description HTS Heading
HTS Heading/Article Subheading description Base Rate Staging Category I. CHEMICAL ELEMENTS 2801 Fluorine, chlorine, bromine and iodine: 28011000 -Chlorine Free E 28012000 -Iodine Free E 280130 -Fluorine; bromine: 28013010 --Fluorine 3.7% A 28013020 --Bromine 5.5% A 28020000 Sulfur, sublimed or precipitated; colloidal sulfur Free E Carbon (carbon blacks and other forms of carbon not 28030000 elsewhere specified or included) Free E 2804 Hydrogen, rare gases and other nonmetals: 28041000 -Hydrogen 3.7% A -Rare gases: 28042100 --Argon 3.7% A 28042900 --Other 3.7% A 28043000 -Nitrogen 3.7% A 28044000 -Oxygen 3.7% A 28045000 -Boron; tellurium Free E -Silicon: --Containing by weight not less than 99.99 percent of 28046100 silicon Free E 280469 --Other: ---Containing by weight less than 99.99 percent but not 28046910 less than 99 percent of silicon 5.3% B 28046950 ---Other 5.5% B 28047000 -Phosphorus Free E 28048000 -Arsenic Free E 28049000 -Selenium Free E Alkali or alkaline-earth metals; rare-earth metals, scandium and yttrium, whether or not intermixed or 2805 interalloyed; mercury: -Alkali metals: 28051100 --Sodium 5.3% B 28051900 --Other 5.5% B -Alkaline-earth metals: 28052100 --Calcium 3% A 280522 --Strontium and barium: 28052210 ---Strontium 3.7% A 28052220 ---Barium Free E -Rare-earth metals, scandium and yttrium, whether or 28053000 not intermixed or interalloyed 5% A 28054000 -Mercury 1.7% A II. INORGANIC ACIDS AND INORGANIC OXYGEN COMPOUNDS OF NONMETALS Hydrogen chloride (Hydrochloric acid); chlorosulfuric 2806 acid: 28061000 -Hydrogen -

Rhenium Trioxide, Rhenium Heptoxide and Rehnium Trichloride As Hydrogenation Catalysts
Brigham Young University BYU ScholarsArchive Theses and Dissertations 1958-07-21 Rhenium trioxide, rhenium heptoxide and rehnium trichloride as hydrogenation catalysts William J. Bartley Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd BYU ScholarsArchive Citation Bartley, William J., "Rhenium trioxide, rhenium heptoxide and rehnium trichloride as hydrogenation catalysts" (1958). Theses and Dissertations. 8161. https://scholarsarchive.byu.edu/etd/8161 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. (QJ> I ,_t;z .1537 {1~ c~z- RHENIUMTRIOXIDE, RHENIUMHEPTOXIDE AND RHENIUMTRICHLORIDE AS HYDROGENATIONCATALYSTS A Thesis Submitted to the Department of Chemistry Brigham Young University Provo, Utah In Partial Fulfillment Of the Requirements for the Degree of Master of Science by William J. Bartley August, 1958 This thesis by William J. Bartley is accepted in its present form by the Department of Chemistry of the Brigham Young University as satisfying the thesis requirements for the degree of Master of Science. ii ACKNOWLEDGEMENTAND DEDICATION 'fhe author wishes to express his sincere appreciation and thanks to all who have assisted in any way toward the com- pletion of this work. Special thanks is extended to Dr. H. Smith Broadbent, who gave freely of his kind assistance and excellent advice. To my parents, Mr. and Mrs. Wm. Bartley, goes my grati- tude and appreciation for their undeviating support. My love and devotion go to my wonderful wife, Earlene, for her patience and encouragement. -

Heterogeneous Catalysis for the Upgrading of Biomass Derived Chemicals Via Hydrodeoxygenation Elizabeth Barrow
University of South Carolina Scholar Commons Theses and Dissertations Spring 2019 Heterogeneous Catalysis for the Upgrading of Biomass Derived Chemicals via Hydrodeoxygenation Elizabeth Barrow Follow this and additional works at: https://scholarcommons.sc.edu/etd Part of the Chemical Engineering Commons Recommended Citation Barrow, E.(2019). Heterogeneous Catalysis for the Upgrading of Biomass Derived Chemicals via Hydrodeoxygenation. (Doctoral dissertation). Retrieved from https://scholarcommons.sc.edu/etd/5294 This Open Access Dissertation is brought to you by Scholar Commons. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. HETEROGENEOUS CATALYSIS FOR THE UPGRADING OF BIOMASS DERIVED CHEMICALS VIA HYDRODEOXYGENATION by Elizabeth Barrow Bachelor of Science Charleston Southern University, 1998 Master of Science University of Miami, 2000 Submitted in Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy in Chemical Engineering College of Engineering and Computing University of South Carolina 2019 Accepted by: Jochen Lauterbach, Major Professor Andreas Heyden, Committee Member John Regalbuto, Committee Member Donna Chen, Committee Member John Weidner, Committee Member Cheryl L. Addy, Vice Provost and Dean of the Graduate School © Copyright by Elizabeth Barrow, 2019 All Rights Reserved. ii DEDICATION This work is dedicated to my boys, Shedrick and William. May they see the importance of education, the benefit of hard work, and the strength that comes from a family’s love. iii ACKNOWLEDGEMENT I am extremely grateful to my PhD advisor Jochen Lauterbach. He accepted me into his research group believing that I had potential. Considering that this course of study was a completely new field to me, it could not have been an easy task for him to give me the time and resources to learn to be an engineer. -
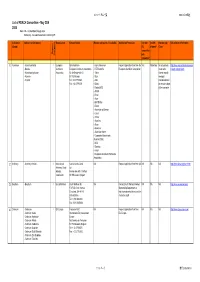
List of REACH Consortium - May 22Th 2008 Note: NA - Not Identified Through Desk Potentially - No Explicit Statement Restricting OR
コンソーシアム一覧 2008/05/22現在 List of REACH Consortium - May 22th 2008 Note: NA - not identified through desk Potentially - no explicit statement restricting OR Substance Substance (Compound) Organisation Contact Details Member companies (if available) Application Procedure Can non- Are OR Membership URL/relevant Information (Group) EU allowed? Costs companies Organic / Inorganic join consortia? 1 Aluminium - Aluminium Metal I European Eirik Nordheim - Hydro Aluminium Request application form from the Yes Potentially No actual data; http://www.eaa.net/en/environmen - Bauxite Aluminium European Aluminium Association - RTZ/Comalco European Aluminium Association costs to be t-health-safety/reach/ - Aluminiumhydroxide Association Av. de Broqueville 12 - Talum shared equally - Alumina B-1150 Brussel - Sual amongst - Cryolite Tel: +32 27756363 - Alba members based Fax: +32 27790531 - Dubal on annual budget - Dadco(AOS) of the consortia - AMAG - Rusal - Aluar - BHP-Billiton - Elkem - Aluminium of Greece - Corus - Trimet - Alcan Inc - Alcoa - Glencore - Aluminium Konin - Companhia Brasileirade Aluminio(CBA) - NLM - Slovalco - Kaiser - European Aluminuim Particulate Association 2 Antimony - Antimony trioxide I International Karine Van de Velde NA Request application form from i2A NA NA NA http://www.iaoia.org/reach.htm Antimony Oxide ia2 Industry Avenue des Arts 13 6 floor Association 1210 Brussels, Belgium 3 Beryllium - Beryllium I Brush Wellman Brush Wellman Inc NA Contact Brush Wellman Product NA NA NA http://www.euromines.org/ 17876 St. Clair Avenue -

Analysis of Rhenium in Molybdenites
Circular 108 Analysis of Rhenium in Molybdenites by LORNA M. GOEBEL STATE BUREAU OF MINES AND MINERAL RESOURCES NEW MEXICO INSTITUTE OF MINING AND TEC HNOLOGY CAMPUS STATION SOCORRO, NEW MEXICO THE NEW MEXICO BUREAU OF MINES AND MINERAL RESOURCES Don H. Baker, Jr., Director Full-Time Staff JOYCE M. AGUILAR, Stenographer WILLIAM L. HAWKS, Materials Engineer WILLIAM E. ARNOLD, Scientific Illustrator FRANK E. KOTTLOWSKI, Sr. Geol. & Ass't. Dir. ROSHAN B. BHAPPU, Senior Metallurgist (on lv.) ALEX. NICHOLSON, Geologist-Editor ROBERT A. BIEBERMAN, Petroleum Geologist ROBERT L. PRICE, Draftsman LYNN A. BRANDVOLD, Assistant Chemist JACQUES R. RENAULT, Geologist ELISE BROWER, Assistant Chemist (on lv.) JOHN W. SHOMAKER, Geologist RICHARD R. CHAVEZ, Lab. Assistant JACKIE H. SMITH, Lab. Assistant Lois M. DEVLIN, Office Manager MARILYNN SZYDLOWSKI, Secretary JO DRAKE, Director's Secretary ROBERT H. WEBER, Senior Geologist ROUSSEAU H. FLOWER, Senior Paleontologist SUE WILKS, Typist ROY W. FOSTER, Petroleum Geologist MAX E. WILLARD, Economic Geologist JUARINE W. WOOLDRIDGE, Editorial Clerk Part-Time Staff M A R T H A K . A R N O L D , Editorial Assistant RUFIE MONTOYA, Dup. Mach. Oper. JOHN Gus BLAISDELL, Public Relations J A M E S REICHE, Instrument Manager RONALD A. B R I E R L E Y , Ass't Prof. Biology ROMAN, Research Metallurgist W. KELLY ROBERT LEASE, SUMMERS, Geologist G e o l o g i s t FRANK B. TITUS, Geologist Graduate Students ELISE BROWER, Geochemist CHE-CHEN LIU, Metallurgist SAUL ESCALERA, Metallurgist WALTER H. PIERCE, Geologist WALTER W. FISHER, Metallurgist HMA ROFFMAN, Geochemist MARSHA KOEHN, Geologist DAVID A. -

The Electrodeposition of Rhenium and Its Alloys Grant No
A Final Report on The Electrodeposition of Rhenium and Its Alloys Grant No. FA9550-10-1-0520 Authors Prof. Shelton Ray Taylor, PI College of Technology University of Houston [email protected] Prof. Noam Eliaz, Co-Inv. Faculty of Engineering Tel Aviv University [email protected] Prof. Eliezer Gileadi, Co-Inv. School of Chemistry Tel Aviv University [email protected] Report Date 18-09-2015 Reporting Period Start Date: September 15, 2010 Reporting Period End Date: September 14, 2015 Sponsoring Agency Air Force Office of Scientific Research (AFOSR) 875 Randolph Street Suite 325, Room 3112 Arlington, VA 22203 Program Manager Dr. Ali Sayir Table of Contents Page 1.0 Abstract 2 2.0 Project Summary 5 3.0 Statement of Work 6 4.0 Significant Outcomes 7 4.1 Mechanistic findings from process optimization studies 7 4.1.1 The electrodeposition of Re-M alloys 7 4.1.2 The effect of bath additives, potentiostatic plating and pulse plating 13 4.1.3 The initial stages of Re-Ni electrodeposition (0.05–60s) 14 4.1.4 Atomic scale characterization 19 4.1.5 Electrodeposition of Re-Ir-base alloys 19 4.1.6 Electroless deposition of Re-Co and Re-Fe alloys 20 4.2 Mechanistic understanding of Re and Re-M electrodeposition 21 4.2.1 Electrochemical Investigation 21 4.2.1.1 Interaction between the citric acid and copper electrode. 22 − 4.2.1.2 The interaction between citric acid, Ni(II) and ReO4 25 4.2.1.3 Mechanism of Ni(II) reduction 27 4.2.1.4 Reduction mechanism of NH4ReO4 31 4.2.1.5 Proposed mechanism for Re–Ni electro-deposition 33 4.2.2 Mechanistic understanding