Approaches and Limitations of Species Level Diagnostics in Flowering Plants
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Natural Heritage Program List of Rare Plant Species of North Carolina 2016
Natural Heritage Program List of Rare Plant Species of North Carolina 2016 Revised February 24, 2017 Compiled by Laura Gadd Robinson, Botanist John T. Finnegan, Information Systems Manager North Carolina Natural Heritage Program N.C. Department of Natural and Cultural Resources Raleigh, NC 27699-1651 www.ncnhp.org C ur Alleghany rit Ashe Northampton Gates C uc Surry am k Stokes P d Rockingham Caswell Person Vance Warren a e P s n Hertford e qu Chowan r Granville q ot ui a Mountains Watauga Halifax m nk an Wilkes Yadkin s Mitchell Avery Forsyth Orange Guilford Franklin Bertie Alamance Durham Nash Yancey Alexander Madison Caldwell Davie Edgecombe Washington Tyrrell Iredell Martin Dare Burke Davidson Wake McDowell Randolph Chatham Wilson Buncombe Catawba Rowan Beaufort Haywood Pitt Swain Hyde Lee Lincoln Greene Rutherford Johnston Graham Henderson Jackson Cabarrus Montgomery Harnett Cleveland Wayne Polk Gaston Stanly Cherokee Macon Transylvania Lenoir Mecklenburg Moore Clay Pamlico Hoke Union d Cumberland Jones Anson on Sampson hm Duplin ic Craven Piedmont R nd tla Onslow Carteret co S Robeson Bladen Pender Sandhills Columbus New Hanover Tidewater Coastal Plain Brunswick THE COUNTIES AND PHYSIOGRAPHIC PROVINCES OF NORTH CAROLINA Natural Heritage Program List of Rare Plant Species of North Carolina 2016 Compiled by Laura Gadd Robinson, Botanist John T. Finnegan, Information Systems Manager North Carolina Natural Heritage Program N.C. Department of Natural and Cultural Resources Raleigh, NC 27699-1651 www.ncnhp.org This list is dynamic and is revised frequently as new data become available. New species are added to the list, and others are dropped from the list as appropriate. -

EDGG Event Report on the International Symposium On
14 Palaearctic Grasslands 43 October 2019) EDGG event DOI: 10.21570/EDGG.PG.43.14-18 Report on the International Symposium on Grassland Ecology and Conservation in Hohhot, Inner Mongolia, China An International Symposium on Grassland Ecology and Con- activities promoted by the EDGG. Many of the participants servation took place in Hohhot, at the Inner Mongolia Uni- expressed an interest in the GrassPlot database and in the versity (IMU), on August 21–23, 2019. It was jointly organ- EDGG activities. It was agreed that invitations to join the ised by the School of Ecology and Environment of the IMU EDGG will be sent out in order to support data sharing and and by regional branches of the China Association for Sci- to encourage new researchers from the West and Central ence and Technology, under the auspices of the Chinese Asian steppe regions to join the network. Grassland Society, the Ecological Society of China and the In the afternoon of the second day symposium participants Inner Mongolia Grassland Association. had the opportunity to visit two leading companies in their The main topic of the Symposium was modelling, monitor- respective sectors: the Mengniu Dairy Company Ltd. and the ing, sustainable use, conservation and management of Mengcao Eco-environment Company Ltd. grasslands (with special regard to natural steppes) through The first is one of the leading dairy product manufacturers transdisciplinary and transnational cooperation. The Sympo- in China, with an annual production capacity of 10.27 mil- sium included two keynote lectures and 40 talks and it was lion tons (as of June 2019, http://www.mengniuir.com/ attended by 228 participants from seven countries, plus a html/about_profile.php); the second specializes in "grass, number of students from the IMU (Figs. -

David A. Rasmussen, 2 Elena M. Kramer, 3 and Elizabeth A. Zimmer 4
American Journal of Botany 96(1): 96–109. 2009. O NE SIZE FITS ALL? M OLECULAR EVIDENCE FOR A COMMONLY INHERITED PETAL IDENTITY PROGRAM IN RANUNCULALES 1 David A. Rasmussen, 2 Elena M. Kramer, 3 and Elizabeth A. Zimmer 4 Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, Massachusetts 02138 USA Petaloid organs are a major component of the fl oral diversity observed across nearly all major clades of angiosperms. The vari- able morphology and development of these organs has led to the hypothesis that they are not homologous but, rather, have evolved multiple times. A particularly notable example of petal diversity, and potential homoplasy, is found within the order Ranunculales, exemplifi ed by families such as Ranunculaceae, Berberidaceae, and Papaveraceae. To investigate the molecular basis of petal identity in Ranunculales, we used a combination of molecular phylogenetics and gene expression analysis to characterize APETALA3 (AP3 ) and PISTILLATA (PI ) homologs from a total of 13 representative genera of the order. One of the most striking results of this study is that expression of orthologs of a single AP3 lineage is consistently petal-specifi c across both Ranunculaceae and Berberidaceae. We conclude from this fi nding that these supposedly homoplastic petals in fact share a developmental genetic program that appears to have been present in the common ancestor of the two families. We discuss the implications of this type of molecular data for long-held typological defi nitions of petals and, more broadly, the evolution of petaloid organs across the angiosperms. Key words: APETALA3 ; MADS box genes; petal evolution; PISTILLATA ; Ranunculales. -

PHCOG RES.: Research Article Essential Oil Composition of the Dracocephalum Moldavica L from Xinjiang in China
[Downloaded free from http://www.phcogres.com on Friday, June 25, 2021, IP: 249.80.203.154] Pharmacognosy Research [Phcog Res.] Vol 1, Issue 4, Jul-Aug, 2009 Page 172-174 (An Official Publication of Pharmacognosy Network Worldwide) Received: 18 May, 2009 Modified: 02 April, 2009 Accepted: 03 June, 2009 PHCOG RES.: Research Article Essential Oil Composition of the Dracocephalum moldavica L from Xinjiang in China Tian Shugea*, Zhou Xiaoyingb, Zhang Fana, An Dongqinga, Yang Taoc a* College of TCM, XinJiang Medical University, Urumqi-830011, XinJiang, China b College of Pharmacy, XinJiang Medical University, Urumqi-830011, XinJiang, China c Institute of Quality Testing of Xinjiang; Urumqi-830002, XinJiang, China E.mail: [email protected] ABSTRACT The essential oil of Dracocephalum moldavica L from Xinjiang in China was isolated by hydrodistillation in yield of 0.15 %(w/w). The chemical composition of the essential oil was analyzed by GC and GCMS. Fifty-one compounds accounting for 99.45% of the total oil were identified. The major components wereα-Citral (32.55%),β-Citral (23.53%), Acetic acid, geranial ester(21.32%), Trans-Geraniol(3.38%), Nerolacetate(3.38%), Octane(2.14%), and 2,4,6-Trimethyl -3-cyclohexene-1-carboxaldehyde (1.3%). Monotenepers were the main group of compounds. Keywords: Dracocephalum moldavica L, essential oil composition, GC-MS. INTRODUCTION during flowering. Voucher specimens were deposited in Traditional Chinese Medicine College Museum of Chinese Dracocephalum moldavica L. is a perennial herb belonging herbal samples of Xinjiang Medical University. to the Lamiaceae (Labiatae) family. is an annual herbaceous aromatic plant belonging to the family Lamiaceae (Labiatae). -

(Dr. Sc. Nat.) Vorgelegt Der Mathematisch-Naturwissenschaftl
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2012 Flowers, sex, and diversity: Reproductive-ecological and macro-evolutionary aspects of floral variation in the Primrose family, Primulaceae de Vos, Jurriaan Michiel Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-88785 Dissertation Originally published at: de Vos, Jurriaan Michiel. Flowers, sex, and diversity: Reproductive-ecological and macro-evolutionary aspects of floral variation in the Primrose family, Primulaceae. 2012, University of Zurich, Facultyof Science. FLOWERS, SEX, AND DIVERSITY. REPRODUCTIVE-ECOLOGICAL AND MACRO-EVOLUTIONARY ASPECTS OF FLORAL VARIATION IN THE PRIMROSE FAMILY, PRIMULACEAE Dissertation zur Erlangung der naturwissenschaftlichen Doktorwürde (Dr. sc. nat.) vorgelegt der Mathematisch-naturwissenschaftliche Fakultät der Universität Zürich von Jurriaan Michiel de Vos aus den Niederlanden Promotionskomitee Prof. Dr. Elena Conti (Vorsitz) Prof. Dr. Antony B. Wilson Dr. Colin E. Hughes Zürich, 2013 !!"#$"#%! "#$%&$%'! (! )*'+,,&$-+''*$.! /! '0$#1'2'! 3! "4+1%&5!26!!"#"$%&'(#)$*+,-)(*#! 77! "4+1%&5!226!-*#)$%.)(#!'&*#!/'%#+'.0*$)/)"$1'(12%-).'*3'0")"$*.)4&4'*#' "5*&,)(*#%$4'+(5"$.(3(-%)(*#'$%)".'(#'+%$6(#7.'2$(1$*.".! 89! "4+1%&5!2226!.1%&&'%#+',!&48'%'9,%#)()%)(5":'-*12%$%)(5"'"5%&,%)(*#'*3' )0"';."&3(#!'.4#+$*1"<'(#'0")"$*.)4&*,.'%#+'0*1*.)4&*,.'2$(1$*.".! 93! "4+1%&5!2:6!$"2$*+,-)(5"'(12&(-%)(*#.'*3'0"$=*!%14'(#'0*1*.)4&*,.' 2$(1$*.".>'5%$(%)(*#'+,$(#!'%#)0".(.'%#+'$"2$*+,-)(5"'%..,$%#-"'(#' %&2(#"'"#5($*#1"#).! 7;7! "4+1%&5!:6!204&*!"#")(-'%#%&4.(.'*3'!"#$%&''."-)(*#'!"#$%&''$"5"%&.' $%12%#)'#*#/1*#*204&4'%1*#!'1*2$0*&*!(-%&&4'+(.)(#-)'.2"-(".! 773! "4+1%&5!:26!-*#-&,+(#!'$"1%$=.! 7<(! +"=$#>?&@.&,&$%'! 7<9! "*552"*?*,!:2%+&! 7<3! !!"#$$%&'#""!&(! Es ist ein zentrales Ziel in der Evolutionsbiologie, die Muster der Vielfalt und die Prozesse, die sie erzeugen, zu verstehen. -

Determination of Taxonomic Status of Chinese Species of the Genus Clematis by Using High Performance Liquid Chromatography–Mass Spectrometry (Hplc-Ms) Technique
Pak. J. Bot., 42(2): 691-702, 2010. DETERMINATION OF TAXONOMIC STATUS OF CHINESE SPECIES OF THE GENUS CLEMATIS BY USING HIGH PERFORMANCE LIQUID CHROMATOGRAPHY–MASS SPECTROMETRY (HPLC-MS) TECHNIQUE MUHAMMAD ISHTIAQ CH1, 2*, Q. HE2, S. FENG, YI WANG2, P.G. XIAO2, YIYU CHENG2* AND HABIB AHMED3 1Department of Botany, Mirpur University of Science & Technology (MUST) Bhimber Campus, (AK) Pakistan 2Department of Chinese Medicine Science and Engineering, College of Pharmaceutical Sciences, Institute of Bioinformatics, Zhejiang University, No. 38 Zhe da Road Hangzhou, 310027, P. R. China 3Department of Botany, University of Hazara, Garden Campus Mansehra, NWFP, Pakistan. Abstract A comparative taxonomic study using chemometric and numerical taxonomic approaches on 15 populations of 12 species of major taxa of the genus Clematis, belonging to sections, Rectae, Clematis, Meclatis, Tubulosae and Viorna were analyzed by HPLC coupled with diode array detector and ESI-MS. The chemodiversity profile of saponins proved to be useful taxonomic markers for the genus and results are presented in phenograms. The compound ‘Huzhangoside D’ was the most abundant in analyzed species of the genus. Numerical taxonomic study was also conducted based on phylogenetically informative characters which corroborated with chemical fingerprinting findings. The significance of chemical markers in taxonomic study as well as their correlation between morphology and chemical compound profile is also debated with its significant role in botanic drugs identification. Introduction Clematis L., is the second largest genus of Ranunculaceae, which comprises of more than 300 species worldwide and among these 147 (93 endemic) species are found in China (Wang, 1999). Traditional classification systems are usually based on floral and vegetative characters, and have been used for taxonomic divisions of many plants. -

Mechanical Architecture and Development in Clematis
Research MechanicalBlackwell Publishing Ltd. architecture and development in Clematis: implications for canalised evolution of growth forms S. Isnard1, T. Speck2 and N. P. Rowe1 1Botanique et Bioinformatique de l’Architecture des Plantes, UMR 5120 CNRS, TA40/PS2, Boulevard de la Lironde, 34398 Montpellier, France; 2Plant Biomechanics Group, Institute for Biology II, Botanical Garden of the Albert-Ludwigs-Universität, Schänzlestrasse 1, D-79104 Freiburg, Germany Summary Author for correspondence: • Mechanical architectures of two Clematis species, the herbaceous perennial S. Isnard Clematis recta and the woody liana, Clematis vitalba, were investigated and + Tel: 33 (0) 467617553 compared with the woody rhizomatous sand dune plant Clematis flammula var. Fax: +33 (0) 467615668 Email: [email protected] maritima. • Bending mechanical properties of stems from various developmental stages were Received: 25 September 2002 compared and related to stem geometry and relative proportions of tissues during Accepted: 28 February 2003 development. doi: 10.1046/j.1469-8137.2003.00771.x • Clematis vitalba and C. flammula var. maritima showed mechanical architectures with reductions in structural Young’s modulus of the stem during ontogeny. Irrever- sible loss of stem rigidity was mediated by disruption, separation and eventual loss of primary phloem fibres via secondary growth of the periderm and cambial activity. Each species showed variations of non-self-supporting mechanical architecture relating to specific habitat preferences. In aerial stems of C. recta the structural Young’s modulus remained approximately constant during ontogeny, a mechanical signal characteristic for semi-self-supporting architectures. •Woody aerial plant stems are extremely rare in the Ranunculaceae and seldom, if ever, show self-supporting characteristics. -
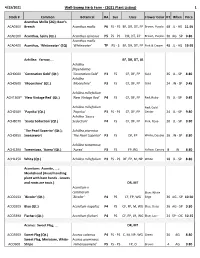
WSHF Catalog
4/23/2021 Well-Sweep Herb Farm - (2021 Plant Listing) 1 Stock # Common Botanical HA Sun Uses Flower ColorHT When Price Acanthus Mollis (2Q); Bear's ACA030X Breech Acanthus mollis P6 FS - PS BF, DR, DT, FP Brown, Purple- 48 JL - AG 11.95 ACA010X Acanthus, Spiny (Qt.) Acanthus spinosus P5 FS - PS DR, DT, FP Brown, Purple- 30 AG - SP 9.80 Acanthus mollis ACA040X Acanthus, `Whitewater' (2Q) `Whitewater' TP PS - S BF, DR, DT, FP Pink & Cream 48 JL - AG 19.95 Achillea: Yarrow, ... BF, DR, DT, LB Achillea filipendulina ACH000X `Coronation Gold' (Qt.) `Coronation Gold' P3 FS CF, DF, FP Gold 36 JL - SP 8.80 Achillea ACH050X `Moonshine' (Qt.) `Moonshine' P3 FS CF, DF, FP Gold 24 JN - SP 9.45 Achillea millefolium ACH130X* `New Vintage Red' (Qt.) `New Vintage Red' P4 FS CF, DF, FP Red, Ruby- 15 JL - SP 9.45 Achillea millefolium Red; Gold ACH250X `Paprika' (Qt.) `Paprika' P3 FS - PS CF, DF, FP Center 24 JL - SP 9.80 Achillea `Saucy ACH807X `Saucy Seduction' (Qt.) Seduction' P4 FS CF, DF, FP Pink, Rose- 20 JL - SP 9.80 `The Pearl Superior' (Qt.); Achillea ptarmica ACH095X Sneezewort `The Pearl Superior' P3 FS DF, FP White; Double 16 JN - SP 8.80 Achillea tomentosa ACH120X Tomentosa, `Aurea' (Qt.) `Aurea' P3 FS FP, RG Yellow, Canary- 8 JN 8.80 ACH125X White (Qt.) Achillea millefolium P3 FS - PS DF, FP, M, NP White 18 JL - SP 8.80 Aconitum: Aconite, ... ; Monkshood (Avoid handling plant with bare hands - Leaves and roots are toxic.) DR, MT Aconitum x cammarum Blue; White ACO015X `Bicolor' (Qt.) `Bicolor' P4 PS CF, FP, WG Edge 36 AG - SP 10.50 ACO020X Blue (Qt.) Aconitum napellus P4 PS CF, FP, M, WG Blue, Deep- 36 AG - SP 9.80 ACO339X Fischeri (Qt.) Aconitum fischeri P4 PS CF, FP, LB, WG Blue, Lav.- 24 SP - OC 10.15 Acorus: Sweet Flag, .. -

Lamiaceae) Species from Iran Alireza Dolatyari & Asghar Kamrani
Wulfenia 22 (2015): xxx–xxx Mitteilungen des Kärntner Botanikzentrums Klagenfurt Chromosome number and morphology of some accessions of four Lallemantia Fisch. & C.A. Mey. (Lamiaceae) species from Iran Alireza Dolatyari & Asghar Kamrani Summary: Lallemantia is a small genus comprising only five species. The upper corolla lip with two internal longitudinal folds differentiates the genus from closely related taxa. Uniform chromosome number and ploidy level have already been reported for the genus. In order to provide chromosomal data and to trace chromosome number changes both at inter- and intra-specific levels[Referee: Why do you expect any chromosome number changes?], we studied 10 accessions of four Lallemantia species from Iran. In accordance with previous results, we found 2n = 2x = 14 for all the accessions studied; B-chromosomes were only found in one accession of L. iberica. In addition, detailed karyotypic data are presented for each accession. The mean value of centromere indices, 0.398, demonstrated the symmetric karyotype for the whole genus. Altogether, our data indicate that Lallemantia is a karyologically stable genus. Keywords: B-chromosome, karyotype, Lallemantia, life form, Nepetoideae The mint family (Lamiaceae) has a world-wide distribution and includes 7200 species (Bräuchler et al. 2010), many of which are aromatic and medicinal. Jamzad (2012) reported from Iran 46 genera in 5 subfamilies: Ajugoideae Kostel., Lamioideae Harley, Nepetoideae (Dumort.) Luerss., Scutellarioideae (Dumort.) Caruel and Viticoideae Briq. The Nepetoideae subfamily is the largest, including 27 out of 46 Iranian genera and it is characterized by the presence of hexacolpate three-nucleate pollens (Cantino & Sanders 1986) as well as the presence of rosmarinic acid (Harley et al. -

Does the Arcto-Tertiary Biogeographic Hypothesis Explain the Disjunct Distribution of Northern Hemisphere Herbaceous Plants? the Case of Meehania (Lamiaceae)
RESEARCH ARTICLE Does the Arcto-Tertiary Biogeographic Hypothesis Explain the Disjunct Distribution of Northern Hemisphere Herbaceous Plants? The Case of Meehania (Lamiaceae) Tao Deng1,2,3, Ze-Long Nie4, Bryan T. Drew5, Sergei Volis2, Changkyun Kim2, Chun-Lei Xiang2, Jian-Wen Zhang2, Yue-Hua Wang1*, Hang Sun2* a11111 1 School of Life Science, Yunnan University, Kunming, Yunnan, China, 2 Key Laboratory for Plant Diversity and Biogeography of East Asia, Chinese Academy of Sciences, Kunming, Yunnan, China, 3 University of Chinese Academy of Sciences, Beijing, China, 4 Key Laboratory of Plant Resources Conservation and Utilization, College of Biology and Environmental Sciences, Jishou University, Jishou, Hunan, China, 5 Department of Biology, University of Nebraska at Kearney, Kearney, Nebraska, United States of America * [email protected] (HS); [email protected] (YHW) OPEN ACCESS Citation: Deng T, Nie Z-L, Drew BT, Volis S, Kim C, Abstract Xiang C-L, et al. (2015) Does the Arcto-Tertiary Biogeographic Hypothesis Explain the Disjunct Despite considerable progress, many details regarding the evolution of the Arcto-Tertiary Distribution of Northern Hemisphere Herbaceous flora, including the timing, direction, and relative importance of migration routes in the evolu- Meehania Plants? The Case of (Lamiaceae). PLoS tion of woody and herbaceous taxa of the Northern Hemisphere, remain poorly understood. ONE 10(2): e0117171. doi:10.1371/journal. pone.0117171 Meehania (Lamiaceae) comprises seven species and five subspecies of annual or perenni- al herbs, and is one of the few Lamiaceae genera known to have an exclusively disjunct dis- Academic Editor: Qi Wang, Institute of Botany, CHINA tribution between eastern Asia and eastern North America. -

2018 Tilth Alliance May Edible Plant Sale Herb and Flower Plant List
2018 Tilth Alliance May Edible Plant Sale Herb and Flower Plant List Updated 4/30/2018 Please be aware that we may not have all the varieties listed in this document at the 2018 May Edible Plant Sale. Occasionally, the growers will experience a crop failure or the plants will be too small to sell. We apologize for any inconvenience this may cause. All our herb and flower starts are organically grown and/or Certified Organic by Cascade Cuts in Bellingham, Rent's Due Ranch in Stanwood, WA, Sunseed Farms in Acme, WA, Growing Garden in Bellingham, WA and The Root Cellar in Yelm, WA. Edible Flowers Alyssum Mixed Colors Lobularia maritima. You may know that alyssum has a light honey-like fragrance and that it blooms non-stop all summer with little care but have you ever tasted Alyssum? Well it doesn't taste strong but it sure looks pretty decorating summer cakes, fruit salad and a bowl of ice cream! The young leaves, stems and flowers are all edible plus they attract all kinds of beneficial insects to the garden. How can you go wrong? Grows to 6" tall and spreads up to a foot. Borage Borago officinalis The bright blue, star-shaped flowers (which bloom most of the summer) make borage one of the prettiest herb plants. The leaves are large and leathery and covered in soft spikes. The flavor of the leaves and flowers resembles that of cucumber. The plant will grow to a height of about 18 inches, and spread about 12 inches. Beloved by bumble bees. -

Vojtech Holubec, Czech Republic 2016/2017 Vojtěch Holubec
Wild Seeds Primula agleniana Vojtech Holubec, Czech Republic 2016/2017 Vojtěch Holubec Bazantni 1217/5, CZ-165 00 Praha 6, Czech Republic phone: +420 731 587 826 e-mail: [email protected] November, 2016 Dear rock-garden friends, Welcome to the 24th seed list 2016/17. The seeds were collected mainly in Tibet, Pamir, Alai, Tien Shan, Yunnan, Kamchatka, Sechuan, Kunlun, Karakoram, Sakhalin, Patagonia, Turkey and others. Selected pictures are in my photogallery http://holubec.wbs.cz/ . There is a lot of items that were regenerated in the garden. They are as valuable as the natural collections and can be collected exactly when become ripe. These species proved to be growable in rock gardens in Central Europe. Those items are clearly marked with ex. Some items come from other collectors: Jiri Papousek (JP), Zdenek Obrdlik (ZO) and Jaro Horacek (JH). The plants were determined according to Floras, in case of the recent collections Fl. China, Fl Tajikistan, Fl Kyrgyzstan, Key to the vascular plants of Kamchatka. Some plants were not seen in flowers and therefore it is not possible to guarantee all determination. From previous expeditions there are still available many good items. All seeds are marked with a collecting year. Older ones were stored in refrigerator and they keep a good germination ability (some of them germinate even better the second year). Several abbreviations were used in descriptions: pl- plant, lv, lvs-leaves, fl, fls-flowers, infl-inflorescence. Please, order by both numbers and names to avoid mistakes. Seeds from another locality will be sent when the ordered item is gone.