Comparing Many-Body Approaches Against the Helium Atom Exact Solution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
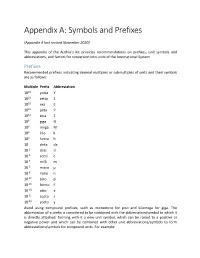
Appendix A: Symbols and Prefixes
Appendix A: Symbols and Prefixes (Appendix A last revised November 2020) This appendix of the Author's Kit provides recommendations on prefixes, unit symbols and abbreviations, and factors for conversion into units of the International System. Prefixes Recommended prefixes indicating decimal multiples or submultiples of units and their symbols are as follows: Multiple Prefix Abbreviation 1024 yotta Y 1021 zetta Z 1018 exa E 1015 peta P 1012 tera T 109 giga G 106 mega M 103 kilo k 102 hecto h 10 deka da 10-1 deci d 10-2 centi c 10-3 milli m 10-6 micro μ 10-9 nano n 10-12 pico p 10-15 femto f 10-18 atto a 10-21 zepto z 10-24 yocto y Avoid using compound prefixes, such as micromicro for pico and kilomega for giga. The abbreviation of a prefix is considered to be combined with the abbreviation/symbol to which it is directly attached, forming with it a new unit symbol, which can be raised to a positive or negative power and which can be combined with other unit abbreviations/symbols to form abbreviations/symbols for compound units. For example: 1 cm3 = (10-2 m)3 = 10-6 m3 1 μs-1 = (10-6 s)-1 = 106 s-1 1 mm2/s = (10-3 m)2/s = 10-6 m2/s Abbreviations and Symbols Whenever possible, avoid using abbreviations and symbols in paragraph text; however, when it is deemed necessary to use such, define all but the most common at first use. The following is a recommended list of abbreviations/symbols for some important units. -

Solving the Schrödinger Equation for Helium Atom and Its Isoelectronic
THE JOURNAL OF CHEMICAL PHYSICS 127, 224104 ͑2007͒ Solving the Schrödinger equation for helium atom and its isoelectronic ions with the free iterative complement interaction „ICI… method ͒ Hiroyuki Nakashima and Hiroshi Nakatsujia Quantum Chemistry Research Institute, Kyodai Katsura Venture Plaza 106, Goryo Oohara 1-36, Nishikyo-ku, Kyoto 615-8245, Japan and Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan ͑Received 31 July 2007; accepted 2 October 2007; published online 11 December 2007͒ The Schrödinger equation was solved very accurately for helium atom and its isoelectronic ions ͑Z=1–10͒ with the free iterative complement interaction ͑ICI͒ method followed by the variational principle. We obtained highly accurate wave functions and energies of helium atom and its isoelectronic ions. For helium, the calculated energy was −2.903 724 377 034 119 598 311 159 245 194 404 446 696 905 37 a.u., correct over 40 digit accuracy, and for H−,itwas−0.527 751 016 544 377 196 590 814 566 747 511 383 045 02 a.u. These results prove numerically that with the free ICI method, we can calculate the solutions of the Schrödinger equation as accurately as one desires. We examined several types of scaling function g and initial function 0 of the free ICI method. The performance was good when logarithm functions were used in the initial function because the logarithm function is physically essential for three-particle collision area. The best performance was obtained when we introduce a new logarithm function containing not only r1 and r2 but also r12 in the same logarithm function. -

Many-Electron System I – Helium Atom and Pauli Exclusion Principle
Lecture 17 Many-electron System I { Helium Atom and Pauli Exclusion Principle Study Goal of This Lecture • Helium atom - many electron wavefunctions • Helium atom - variational ground state (screening) • Eigenstates of a two-spin system 17.1 Helium Atom Helium atom includes two electrons moving around a fixed nucleus with charge Z = 2, we can write down the Hamiltonian as: 2 2 2 2 ^ ~ 2 2 1 Ze Ze e H = − (r1 + r2) − ( + − ) 2me 4π0 r1 r2 r12 2 2 2 2 2 ~ 2 1 Ze ~ 2 1 Ze e = − r1 − − − r2 − + 2me 4π0 r1 2me 4π0 r2 4π0r12 (17.1) | {z } | {z } | {z } H^1 H^2 H^12 = H^1 + H^2 + H^12: 1 If H^12 = 0(or neglected) then the problem is exactly solved. Recall that for a total system composes of independent sub-systems H^T = H^1 + H^2 + ··· ; (17.2) and we can firstly solve all ^ n n n Hnφm = Emφm; (17.3) then the solution of H^T is the product states Y n = φ : (17.4) n If consider as many-electrons, i.e. H^n for the n-th electron. Then the product solu- tion is a natural "independent electron" solution. Even when the electron-electron interactions are non-zero, we will see the independent electorn approximation is a good starting point. Let's consider the Helium atom, we know: H^1φ1 = E1φ1; (17.5) H^2φ2 = E2φ2; and total E = E1 + E2. φ1; φ2 are Helium hydrogen-like atomic orbitals. We know the two ground states (neglect spin for a moment.) H^1φ1s(1) = E1sφ1s(1); (17.6) H^2φ1s(2) = E1sφ1s(2); number 1 and 2 denotes electorn 1 and electron 2 respectively. -

Helium Atom, Approximate Methods
Helium Atom, Approximate Methods 22nd April 2008 I. The Helium Atom and Variational Principle: Approximation Methods for Complex Atomic Systems The hydrogen atom wavefunctions and energies, we have seen, are deter- mined as a combination of the various quantum "dynamical" analogues of classical motions (translation, vibration, rotation) and a central-force inter- action (i.e, the Coulomb interaction between an electron and a nucleus). Now, we consider the Helium atom and will see that due to the attendant 3-body problem for which we cannot determine a close-for, first-principles analytic solution, we will have to find recourse in approximate methods. The Helium atom has 2 electrons with coordinates r1 and r2 as well as a single nucleus with coordinate R. The nuclues carries a Z = +2e charge. The Schrodinger equation is: 2 2 2 h¯ 2 h¯ 2 h¯ 2 1 2 (R; r1; r2) + −2M r − 2me r − 2me r ! 2e2 2e2 e2 + (R; r ; r ) = E (R; r ; r ) −4π R r − 4π R r 4π r r 1 2 1 2 o j − 1j o j − 2j o j 1 − 2j! where the symbol "nabla", when squared, is given by: @2 @2 @2 2 = + + r @x2 @y2 @z2 Keep in mind that the R, r, and r represent the Cartesian coordinates of each paticle. This is a 3-body problem and such problems are not solved exactly. Thus, the problem will be reformulated in terms of 2 variables. The first approximations: Mme , fix the nucleous at the origin (R) = 0. Thus, the Schrodinger equation in relative variables is: 1 2 2 2 h¯ 2 2 2e 1 1 e 1 2 (r1; r2) + + (r1; r2) = E (r1; r2) 2me −∇ − r −4πo r1 r2 4πo r2 r1 j − j Recall that the 2, representing the kinetic energy operator, in spherical r polar coordinates is: 1 @ @ 1 @ @ 1 @2 r2 + sinθ + 2 @r 1 @r 2 @θ 1 @θ 2 2 2 r1 1 1 r1sinθ1 1 1 r1sin θ1 @φ1 The Independent Electron Approximation to Solving the Helium Atom Schrodinger Equation If we neglect electron-electron repulsion in the Helium atom problem, we can simplify and solve the effective 2-body problem. -

Guide for the Use of the International System of Units (SI)
Guide for the Use of the International System of Units (SI) m kg s cd SI mol K A NIST Special Publication 811 2008 Edition Ambler Thompson and Barry N. Taylor NIST Special Publication 811 2008 Edition Guide for the Use of the International System of Units (SI) Ambler Thompson Technology Services and Barry N. Taylor Physics Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899 (Supersedes NIST Special Publication 811, 1995 Edition, April 1995) March 2008 U.S. Department of Commerce Carlos M. Gutierrez, Secretary National Institute of Standards and Technology James M. Turner, Acting Director National Institute of Standards and Technology Special Publication 811, 2008 Edition (Supersedes NIST Special Publication 811, April 1995 Edition) Natl. Inst. Stand. Technol. Spec. Publ. 811, 2008 Ed., 85 pages (March 2008; 2nd printing November 2008) CODEN: NSPUE3 Note on 2nd printing: This 2nd printing dated November 2008 of NIST SP811 corrects a number of minor typographical errors present in the 1st printing dated March 2008. Guide for the Use of the International System of Units (SI) Preface The International System of Units, universally abbreviated SI (from the French Le Système International d’Unités), is the modern metric system of measurement. Long the dominant measurement system used in science, the SI is becoming the dominant measurement system used in international commerce. The Omnibus Trade and Competitiveness Act of August 1988 [Public Law (PL) 100-418] changed the name of the National Bureau of Standards (NBS) to the National Institute of Standards and Technology (NIST) and gave to NIST the added task of helping U.S. -

Quantum Physics (UCSD Physics 130)
Quantum Physics (UCSD Physics 130) April 2, 2003 2 Contents 1 Course Summary 17 1.1 Problems with Classical Physics . .... 17 1.2 ThoughtExperimentsonDiffraction . ..... 17 1.3 Probability Amplitudes . 17 1.4 WavePacketsandUncertainty . ..... 18 1.5 Operators........................................ .. 19 1.6 ExpectationValues .................................. .. 19 1.7 Commutators ...................................... 20 1.8 TheSchr¨odingerEquation .. .. .. .. .. .. .. .. .. .. .. .. .... 20 1.9 Eigenfunctions, Eigenvalues and Vector Spaces . ......... 20 1.10 AParticleinaBox .................................... 22 1.11 Piecewise Constant Potentials in One Dimension . ...... 22 1.12 The Harmonic Oscillator in One Dimension . ... 24 1.13 Delta Function Potentials in One Dimension . .... 24 1.14 Harmonic Oscillator Solution with Operators . ...... 25 1.15 MoreFunwithOperators. .. .. .. .. .. .. .. .. .. .. .. .. .... 26 1.16 Two Particles in 3 Dimensions . .. 27 1.17 IdenticalParticles ................................. .... 28 1.18 Some 3D Problems Separable in Cartesian Coordinates . ........ 28 1.19 AngularMomentum.................................. .. 29 1.20 Solutions to the Radial Equation for Constant Potentials . .......... 30 1.21 Hydrogen........................................ .. 30 1.22 Solution of the 3D HO Problem in Spherical Coordinates . ....... 31 1.23 Matrix Representation of Operators and States . ........... 31 1.24 A Study of ℓ =1OperatorsandEigenfunctions . 32 1.25 Spin1/2andother2StateSystems . ...... 33 1.26 Quantum -

HELIUM ATOM R R2 R1 R2 R1
HELIUM ATOM Now that we have treated the Hydrogen like atoms in some detail, we now proceed to discuss the nextsimplest system: the Helium atom. In this situation, we have tow electrons – with coordinates z r1 and r2 – orbiting a nucleus with charge Z = 2 located at the point R. Now, for the hydrogen atom we were able to ignore the motion of the nucleus r2 by transforming to the center of mass. We then obtained a Schrödinger equation for a single R y effective particle – with a reduced mass that was very close to the electron mass – orbiting the x origin. It turns out to be fairly difficult to r1 transform to the center of mass when dealing with three particles, as is the case for Helium. However, because the nucleus is much more massive than either of the two electrons (MNuc ≈ 7000 mel) it is a very good z approximation to assume that the nucleus sits at the center of mass of the atom. In this approximate set of COM coordinates, then, R=0 r2 and the electron coordinates r and r measure 1 2 y the between each electron and the nucleus. Further, we feel justified in separating the motion of the nucleus (which will roughly x r correspond to rigidly translating the COM of the 1 atom) from the relative d the electrons orbiting the nucleus within the COM frame. Thus, in what follows, we focus only on the motion of the electrons and ignore the motion of the nucleus. We will treat the quantum mechanics of multiple particles (1,2,3…) in much the same way as we described multiple dimensions. -

The International System of Units (SI)
NAT'L INST. OF STAND & TECH NIST National Institute of Standards and Technology Technology Administration, U.S. Department of Commerce NIST Special Publication 330 2001 Edition The International System of Units (SI) 4. Barry N. Taylor, Editor r A o o L57 330 2oOI rhe National Institute of Standards and Technology was established in 1988 by Congress to "assist industry in the development of technology . needed to improve product quality, to modernize manufacturing processes, to ensure product reliability . and to facilitate rapid commercialization ... of products based on new scientific discoveries." NIST, originally founded as the National Bureau of Standards in 1901, works to strengthen U.S. industry's competitiveness; advance science and engineering; and improve public health, safety, and the environment. One of the agency's basic functions is to develop, maintain, and retain custody of the national standards of measurement, and provide the means and methods for comparing standards used in science, engineering, manufacturing, commerce, industry, and education with the standards adopted or recognized by the Federal Government. As an agency of the U.S. Commerce Department's Technology Administration, NIST conducts basic and applied research in the physical sciences and engineering, and develops measurement techniques, test methods, standards, and related services. The Institute does generic and precompetitive work on new and advanced technologies. NIST's research facilities are located at Gaithersburg, MD 20899, and at Boulder, CO 80303. -

HELIUM ATOM R R2 R1 R2 R1
5.61 Physical Chemistry 25 Helium Atom page 1 HELIUM ATOM Now that we have treated the Hydrogen like atoms in some detail, we now proceed to discuss the nextsimplest system: the Helium atom. In this situation, we have tow electrons – with coordinates z r1 and r2 – orbiting a nucleus with charge Z = 2 located at the point R. Now, for the hydrogen atom we were able to ignore the motion of the nucleus r2 by transforming to the center of mass. We then obtained a Schrödinger equation for a single R y effective particle – with a reduced mass that was very close to the electron mass – orbiting the x origin. It turns out to be fairly difficult to r1 transform to the center of mass when dealing with three particles, as is the case for Helium. However, because the nucleus is much more massive than either of the two electrons (MNuc ≈ 7000 mel) it is a very good z approximation to assume that the nucleus sits at the center of mass of the atom. In this approximate set of COM coordinates, then, R=0 r2 and the electron coordinates r and r measure 1 2 y the between each electron and the nucleus. Further, we feel justified in separating the motion of the nucleus (which will roughly x r correspond to rigidly translating the COM of the 1 atom) from the relative d the electrons orbiting the nucleus within the COM frame. Thus, in what follows, we focus only on the motion of the electrons and ignore the motion of the nucleus. -

Introduction to Numerical Projects Format for Electronic Delivery Of
Introduction to numerical projects Here follows a brief recipe and recommendation on how to write a report for each project. • Give a short description of the nature of the problem and the eventual numerical methods you have used. • Describe the algorithm you have used and/or developed. Here you may find it con- venient to use pseudocoding. In many cases you can describe the algorithm in the program itself. • Include the source code of your program. Comment your program properly. • If possible, try to find analytic solutions, or known limits in order to test your program when developing the code. • Include your results either in figure form or in a table. Remember to label your results. All tables and figures should have relevant captions and labels on the axes. • Try to evaluate the reliabilty and numerical stability/precision of your results. If pos- sible, include a qualitative and/or quantitative discussion of the numerical stability, eventual loss of precision etc. • Try to give an interpretation of you results in your answers to the problems. • Critique: if possible include your comments and reflections about the exercise, whether you felt you learnt something, ideas for improvements and other thoughts you’ve made when solving the exercise. We wish to keep this course at the interactive level and your comments can help us improve it. We do appreciate your comments. • Try to establish a practice where you log your work at the computerlab. You may find such a logbook very handy at later stages in your work, especially when you don’t properly remember what a previous test version of your program did. -

Photoelectric Effect Prac
Name ___________________. Photoelectric Effect Prac 1. The threshold frequency in a photoelectric experiment is most closely related to the A) brightness of the incident light B) thickness of the photoemissive metal C) area of the photoemissive metal D) work function of the photoemissive metal 2. When a source of dim orange light shines on a photosensitive metal, no photoelectrons are ejected from its surface. What could be done to increase the likelihood of producing photoelectrons? A) Replace the orange light source with a red light source. B) Replace the orange light source with a higher frequency light source. C) Increase the brightness of the orange light source. D) Increase the angle at which the photons of orange light strike the metal. 3. The threshold frequency for a photoemissive surface is hertz. Which color light, if incident upon the surface, may produce photoelectrons? A) blue B) green C) yellow D) red 4. The threshold frequency of a metal surface is in the violet light region. What type of radiation will cause photoelectrons to be emitted from the metal's surface? A) infrared light B) red light C) ultraviolet light D) radio waves 5. Photons with an energy of 7.9 electron-volts strike a zinc plate, causing the emission of photoelectrons with a maximum kinetic energy of 4.0 electronvolts. The work function of the zinc plate is A) 11.9 eV B) 7.9 eV C) 3.9 eV D) 4.0 eV 6. The threshold frequency for a photoemissive surface is 1.0 × 1014 hertz. What is the work function of the surface? A) 1.0 × 10–14 J B) 6.6 × 10–20 J C) 6.6 × 10–48 J D) 2.2 × 10–28 J 7. -

9081872 Physics Jan. 01
The University of the State of New York REGENTS HIGH SCHOOL EXAMINATION PHYSICS Tuesday, January 23, 2001 — 9:15 a.m. to 12:15 p.m., only The answer paper is stapled in the center of this examination booklet. Open the examination book- let, carefully remove the answer paper, and close the examination booklet. Then fill in the heading on your answer paper. All of your answers are to be recorded on the separate answer paper. For each question in Part I and Part II, decide which of the choices given is the best answer. Then on the answer paper, in the row of numbers for that question, circle with pencil the number of the choice that you have selected. The sample below is an example of the first step in recording your answers. SAMPLE: 1 2 3 4 If you wish to change an answer, erase your first penciled circle and then circle with pencil the num- ber of the answer you want. After you have completed the examination and you have decided that all of the circled answers represent your best judgment, signal a proctor and turn in all examination material except your answer paper. Then and only then, place an X in ink in each penciled circle. Be sure to mark only one answer with an X in ink for each question. No credit will be given for any question with two or more X’s marked. The sample below indicates how your final choice should be marked with an X in ink. SAMPLE: 1 2 3 4 For questions in Part III, record your answers in accordance with the directions given in the exami- nation booklet.