Enabling Direct Preferential Crystallization in a Stable Racemic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aminophylline Catalog Number A1755 Storage
Aminophylline Catalog Number A1755 Storage Temperature –20 °C Replacement for Catalog Number 216895 CAS RN 317-34-0 Storage/Stability Synonyms: theophylline hemiethylenediamine complex; Aminophylline should be kept tightly closed to prevent 3,7-dihydro-1,3-demethyl-1H-purine-2,6-dione CO2 absorption from the atmosphere, which leads to compound with 1,2-ethanediamine (2:1); formation of theophylline and decreased solubility in 1,2 3 (theophylline)2 • ethylenediamine aqueous solutions. Stock solutions should be protected from light and prevented from contact with Product Description metals.2 Molecular Formula: C7H8N4O2 ·1/2 (C2H8N2) Molecular Weight: 210.3 References 1. The Merck Index, 12th ed., Entry# 485. Aminophylline is a xanthine derivative which is a 2. Martindale: The Extra Pharmacopoeia, 31st ed., combination of theophylline and ethylenediamine that is Reynolds, J. E. F., ed., Royal Pharmaceutical more water soluble than theophylline alone. Society (London, England: 1996), pp. 1651-1652. Aminophylline has been widely used as an inhibitor of 3. Data for Biochemical Research, 3rd ed., Dawson, cAMP phosphodiesterase.3 R. M. C., et al., Oxford University Press (New York, NY: 1986), pp. 316-317. Aminophylline has been shown to limit 4. Pelech, S. L., et al., cAMP analogues inhibit phosphatidylcholine biosynthesis in cultured rat phosphatidylcholine biosynthesis in cultured rat hepatocytes.4 It has been used in studies of acute hepatocytes. J. Biol. Chem., 256(16), 8283-8286 hypoxemia in newborn and older guinea pigs.5 The (1981). effect of various xanthine derivatives, including 5. Crisanti, K. C., and Fewell, J. E., Aminophylline aminophylline, on activation of the cystic fibrosis alters the core temperature response to acute transmembrane conductance regulator (CFTR) chloride hypoxemia in newborn and older guinea pigs. -

Download Product Insert (PDF)
PRODUCT INFORMATION Proxyphylline Item No. 20937 CAS Registry No.: 603-00-9 Formal Name: 3,7-dihydro-7-(2-hydroxypropyl)-1,3- N dimethyl-1H-purine-2,6-dione O N Synonym: NSC 163343 C H N O MF: 10 14 4 3 N FW: 238.2 N Purity: ≥98% O UV/Vis.: λmax: 273, 324 nm Supplied as: A crystalline solid OH Storage: -20°C Stability: As supplied, 2 years from the QC date provided on the Certificate of Analysis, when stored properly Laboratory Procedures Proxyphylline is supplied as a crystalline solid. A stock solution may be made by dissolving the proxyphylline in the solvent of choice. Proxyphylline is soluble in organic solvents such as ethanol, DMSO, and dimethyl formamide (DMF), which should be purged with an inert gas. The solubility of proxyphylline in ethanol is approximately 1 mg/ml and approximately 10 mg/ml in DMSO and DMF. Further dilutions of the stock solution into aqueous buffers or isotonic saline should be made prior to performing biological experiments. Ensure that the residual amount of organic solvent is insignificant, since organic solvents may have physiological effects at low concentrations. Organic solvent-free aqueous solutions of proxyphylline can be prepared by directly dissolving the crystalline solid in aqueous buffers. The solubility of proxyphylline in PBS, pH 7.2, is approximately 1 mg/ml. We do not recommend storing the aqueous solution for more than one day. Description Proxyphylline is a methylxanthine derivative that has bronchodilatory actions.1 It has also been reported 2 to have vasodilatory and cardiac stimulatory effects. -
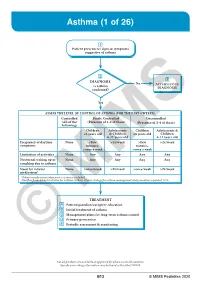
Asthma (1 of 26)
Asthma (1 of 26) 1 Patient presents w/ signs & symptoms suggestive of asthma 2 3 DIAGNOSIS No ALTERNATIVE Is asthma DIAGNOSIS confi rmed? Yes ASSESS THE LEVEL OF CONTROL OF ASTHMA FOR THE PAST 4 WEEKS Controlled Partly Controlled Uncontrolled (All of the (Presence of 1-2 of these) (Presence of 3-4 of these) following) Children Adolescents Children Adolescents & ≤5 years old & Children ≤5 years old Children 6-11 years old 6-11 years old Frequency of daytime None >Few >2x/week >Few >2x/week symptoms minutes, minutes, >once a week >once a week Limitation of activities None Any Any Any Any Nocturnal waking up or None Any Any Any Any coughing due to asthma Need for reliever None >once/week >2x/week >once/week >2x/week medication* *Reliever medications taken prior to exercise excluded. Modified from: Global Initiative for Asthma (GINA). Global strategy for asthma management and prevention: Updated 2020. TREATMENT A Patient/guardian/caregiver education B Initial treatment of asthma C Management plans for long-term asthma control D Primary prevention E © Periodic assessmentMIMS & monitoring Not all products are available or approved for above use in all countries. Specifi c prescribing information may be found in the latest MIMS. B13 © MIMS Pediatrics 2020 Asthma (2 of 26) 1 ASTHMA • A heterogeneous disease w/ chronic infl ammatory disorder of the airways • e most common chronic disease in pediatric age groups that causes signifi cant morbidity • Characterized by history of respiratory symptoms eg wheeze, shortness of breath, chest tightness & cough -

Certificate of Analysis
National Institute of Standards & Technology Certificate of Analysis Standard Reference Material 3254 Green Tea (Camellia sinensis) Leaves This Standard Reference Material (SRM) is intended primarily for use in validating analytical methods for the determination of catechins, xanthines and elements in the leaves of green tea (Camellia sinensis) and similar matrices. SRM 3254 can also be used for quality assurance when assigning values to in-house control materials. This SRM has also been characterized for its DNA sequence. A unit of SRM 3254 consists of five packets, each containing approximately 3 g of leaf powder. The development of SRM 3254 was a collaboration among the National Institute of Standards and Technology (NIST), the National Institutes of Health Office of Dietary Supplements (NIH-ODS), and the Food and Drug Administration Center for Drug Evaluation and Research (FDA CDER). The addition of genetic information was accomplished through collaboration among NIST, NIH-ODS, the U.S. Department of Agriculture (USDA) Agricultural Research Service (ARS), AuthenTechnologies (Richmond, CA), and American Herbal Pharmacopoeia (Scotts Valley, CA). Taxonomic Identification: The taxonomic identity is Camellia sinensis, established through identification by a trained botanist using an herbarium specimen from original material and from associated DNA sequence analysis from botanically authenticated Camellia sinensis. The associated DNA sequences are available in companion FASTA-formatted files [1]. The uncertainty associated with each nucleotide in the sequence, and in turn the uncertainty associated with the DNA sequence as an identifier of species, is expressed in an ordinal scale that represents the confidence estimates of the assigned value (Tables 1 and 2) [2]. These DNA sequences are used as a source of identity data for Camellia sinensis. -

Adenosine Receptor P1 Receptor
Adenosine Receptor P1 receptor Adenosine receptors (ARs) comprise a group of G protein-coupled receptors (GPCR) which mediate the physiological actions of adenosine. To date, four AR subtypes have been cloned and identified in different tissues. These receptors have distinct localization, signal transduction pathways and different means of regulation upon exposure to agonists. A key property of some of Adenosine receptors is their ability to serve as sensors of cellular oxidative stress, which is transmitted by transcription factors, such as NF-κB, to regulate the expression of ARs. The importance of Adenosine receptors in the regulation of normal and pathological processes such as sleep, the development of cancers and in protection against hearing loss will be examined. www.MedChemExpress.com 1 Adenosine Receptor Inhibitors, Agonists, Antagonists, Activators & Modulators (Rac)-Mirabegron D5 5'-N-Ethylcarboxamidoadenosine ((Rac)-YM178 D5) Cat. No.: HY-14773S (NECA) Cat. No.: HY-103173 (Rac)-Mirabegron D5 ((Rac)-YM178 D5) is a 5'-N-Ethylcarboxamidoadenosine (NECA) is a deuterium labeled (Rac)-Mirabegron. nonselective adenosine receptor agonist. (Rac)-Mirabegron is the racemate of Mirabegron. Mirabegron is a selective β3-adrenoceptor agonist. Purity: >98% Purity: 99.86% Clinical Data: No Development Reported Clinical Data: No Development Reported Size: 1 mg, 5 mg Size: 10 mM × 1 mL, 5 mg, 10 mg 8-Cyclopentyl-1,3-dimethylxanthine A2A receptor antagonist 1 Cat. No.: HY-W011955 (CPI-444 analog) Cat. No.: HY-102024 8-Cyclopentyl-1,3-dimethylxanthine (Compound 2a) A2A receptor antagonist 1 (CPI-444 analog) is an is a selective adenosine A1 receptor antagonist antagonist of both adenosine A2A receptor and with Kis of 10.9 nM and 1440 nM for A1 receptor A1 receptor with Ki values of 4 and 264 nM, and A2 receptor, respectively. -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. -

Effects of Aminophylline, Proxyphylline and a Proxy Phylline-Melilotus Extract-Rutin Mixture (Theoesberiven) on the Heart and the Coronary Circulation
EFFECTS OF AMINOPHYLLINE, PROXYPHYLLINE AND A PROXY PHYLLINE-MELILOTUS EXTRACT-RUTIN MIXTURE (THEOESBERIVEN) ON THE HEART AND THE CORONARY CIRCULATION Keisuke TAKEDA, Yumi KATANO, Yoshito NAKAGAWA, Tokumasa TSUKADA, Mikio NAKAZAWA, Takeshi OTORII* and Shoichi IMAI Departmentof Pharmacology,Niigata University School of Medicine, Niigata951, and *Departmentof Pharmacology, YamagataUniversity School of Medicine,Yamagata 990-23, Japan AcceptedJune 16, 1977 Abstract-Effects of aminophylline, proxyphylline and a proxyphylline-Melilotus extract-rutin-mixture (theoesberiven) on the heart and coronary circulation were studied in the canine heart-lung preparation supported by a donor in comparison with those of isoproterenol (Isp). All the three methylxanthines produced an increase in the coronary flow associated with a definite positive inotropic effect. In contrast to the effects of Isp, increase in the heart rate was minimal with these compounds. Myocardial redox potential (JEh) changed to more negative values, although the changes were smaller than that produced by Isp. Since it was found that these substances produced a dis proportionately greater increase in the coronary flow as compared with an increase in the myocardial oxygen consumption, the effects of these substances on the myocardial high-energy phosphate compounds were also studied in the isolated perfused heart preparation of the guinea pig. All the three compounds produced a reduction of the creatine phosphate (Cr-P)/(Cr-Y-1, Inorganic phosphate) ratio and the energy charge of the adenylate system, in the following order, aminophylline; -proxyphylline-theoes beriven. Antagonism towards adenosine was found with the three methylxanthines used. In the isolated left atria] preparation of the guinea-pig, aminophylline induced a definite potentiation of the positive inotropic effect of isoproterenol, while the poten tiation produced by proxyphylline and theoesberiven was minimal. -

Prestwick Collection
(-) -Levobunolol hydrochloride (-)-Adenosine 3',5'-cyclic monophosphate (-)-Cinchonidine (-)-Eseroline fumarate salt (-)-Isoproterenol hydrochloride (-)-MK 801 hydrogen maleate (-)-Quinpirole hydrochloride (+) -Levobunolol hydrochloride (+)-Isoproterenol (+)-bitartrate salt (+,-)-Octopamine hydrochloride (+,-)-Synephrine (±)-Nipecotic acid (1-[(4-Chlorophenyl)phenyl-methyl]-4-methylpiperazine) (cis-) Nanophine (d,l)-Tetrahydroberberine (R) -Naproxen sodium salt (R)-(+)-Atenolol (R)-Propranolol hydrochloride (S)-(-)-Atenolol (S)-(-)-Cycloserine (S)-propranolol hydrochloride 2-Aminobenzenesulfonamide 2-Chloropyrazine 3-Acetamidocoumarin 3-Acetylcoumarin 3-alpha-Hydroxy-5-beta-androstan-17-one 6-Furfurylaminopurine 6-Hydroxytropinone Acacetin Acebutolol hydrochloride Aceclofenac Acemetacin Acenocoumarol Acetaminophen Acetazolamide Acetohexamide Acetopromazine maleate salt Acetylsalicylsalicylic acid Aconitine Acyclovir Adamantamine fumarate Adenosine 5'-monophosphate monohydrate Adiphenine hydrochloride Adrenosterone Ajmalicine hydrochloride Ajmaline Albendazole Alclometasone dipropionate Alcuronium chloride Alexidine dihydrochloride Alfadolone acetate Alfaxalone Alfuzosin hydrochloride Allantoin alpha-Santonin Alprenolol hydrochloride Alprostadil Althiazide Altretamine Alverine citrate salt Ambroxol hydrochloride Amethopterin (R,S) Amidopyrine Amikacin hydrate Amiloride hydrochloride dihydrate Aminocaproic acid Aminohippuric acid Aminophylline Aminopurine, 6-benzyl Amiodarone hydrochloride Amiprilose hydrochloride Amitryptiline hydrochloride -

Nucleosides, Nucleotides, Nucleic Acids and Related Reagents
ホームページではパンフレットに収載されていない化合物,試薬のスケールアップのご用命を受け付けております。 www.tokyokasei.co.jp/jutaku/synthesis Nucleosides, Nucleotides, Nucleic Acids and Related Reagents Genetic information is stored in DNA as combinatorial codes Nucleosides and Their Analogs held in nucleosides and nucleotides, in which form it is passed Nucleosides are glycosylamines made by attaching a from parents to their offspring. Analogs of nucleosides and nucleobase to a ribose or 2’-deoxyribose, which can be nucleotides are used clinically as medicinal agents such as phosphorylated producing nucleotides. Nucleoside analogs reverse transcriptase inhibitors. Therefore, the preparation and are an established class of clinically useful medicinal agents development of these species as effective, selective and possessing a wide range of antiviral and anticancer activities. nontoxic antiviral and antitumor agents has been the subject Consequently, extensive modifications have been made to of intense research.1) both the heterocyclic base and the sugar moiety. Some In addition to this, the development of Polymerase Chain representative examples of these are 9-[(2-hydroxyethoxy) Reaction (PCR) methodology has brought a dramatic change methyl]guanine (acyclovir) developed by Elion in 1977, which and rapid development in studies of DNA. At the current time shows antiviral activity; 3'-azido-3'-deoxythymidine (AZT) the draft version in decoding and mapping human genome discovered by Mitsuya et al. in 1985 and used for the treatment has been almost completed, and the functional analyses of of HIV infection; and cytosine β-D-arabinofuranoside genome and analyses of “Single Nucleotide Polymorphism” (cytarabine) approved by the FDA in 1969 and which has been (SNP) are being vigorously pursued. Discovery of the RNAi shown to display a range anticancer activities. -

2021 Equine Prohibited Substances List
2021 Equine Prohibited Substances List . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s). Prohibited Substances that are identified as Specified Substances in the List below should not in any way be considered less important or less dangerous than other Prohibited Substances. Rather, they are simply substances which are more likely to have been ingested by Horses for a purpose other than the enhancement of sport performance, for example, through a contaminated food substance. LISTED AS SUBSTANCE ACTIVITY BANNED 1-androsterone Anabolic BANNED 3β-Hydroxy-5α-androstan-17-one Anabolic BANNED 4-chlorometatandienone Anabolic BANNED 5α-Androst-2-ene-17one Anabolic BANNED 5α-Androstane-3α, 17α-diol Anabolic BANNED 5α-Androstane-3α, 17β-diol Anabolic BANNED 5α-Androstane-3β, 17α-diol Anabolic BANNED 5α-Androstane-3β, 17β-diol Anabolic BANNED 5β-Androstane-3α, 17β-diol Anabolic BANNED 7α-Hydroxy-DHEA Anabolic BANNED 7β-Hydroxy-DHEA Anabolic BANNED 7-Keto-DHEA Anabolic CONTROLLED 17-Alpha-Hydroxy Progesterone Hormone FEMALES BANNED 17-Alpha-Hydroxy Progesterone Anabolic MALES BANNED 19-Norandrosterone Anabolic BANNED 19-Noretiocholanolone Anabolic BANNED 20-Hydroxyecdysone Anabolic BANNED Δ1-Testosterone Anabolic BANNED Acebutolol Beta blocker BANNED Acefylline Bronchodilator BANNED Acemetacin Non-steroidal anti-inflammatory drug BANNED Acenocoumarol Anticoagulant CONTROLLED Acepromazine Sedative BANNED Acetanilid Analgesic/antipyretic CONTROLLED Acetazolamide Carbonic Anhydrase Inhibitor BANNED Acetohexamide Pancreatic stimulant CONTROLLED Acetominophen (Paracetamol) Analgesic BANNED Acetophenazine Antipsychotic BANNED Acetophenetidin (Phenacetin) Analgesic BANNED Acetylmorphine Narcotic BANNED Adinazolam Anxiolytic BANNED Adiphenine Antispasmodic BANNED Adrafinil Stimulant 1 December 2020, Lausanne, Switzerland 2021 Equine Prohibited Substances List . Prohibited Substances include any other substance with a similar chemical structure or similar biological effect(s). -

Wo 2007/120485 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (43) International Publication Date PCT (10) International Publication Number 25 October 2007 (25.10.2007) WO 2007/120485 A2 (51) International Patent Classification: IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, A61K 31/195 (2006.01) LS, LT, LU, LY,MA, MD, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PG, PH, PL, PT, RO, RS, (21) International Application Number: RU, SC, SD, SE, SG, SK, SL, SM, SV, SY, TJ, TM, TN, PCT/US2007/0081 14 TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW (22) International Filing Date: 30 March 2007 (30.03.2007) (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (25) Filing Language: English GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, TM), (26) Publication Language: English European (AT,BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HU, IE, IS, IT, LT,LU, LV,MC, MT, NL, PL, PT, RO, SE, SI, SK, TR), OAPI (BF, BJ, CF, CG, CI, CM, (30) Priority Data: 60/787,150 30 March 2006 (30.03.2006) US GA, GN, GQ, GW, ML, MR, NE, SN, TD, TG). Declarations under Rule 4.17: (71) Applicant (for all designated States except US): CINER- — as to applicant's entitlement to apply for and be granted a GEN, LLC [US/US]; 146 Medinah Drive, Blue Bell, PA patent (Rule 4.17(U)) 19422-3212 (US). -

Harmonized Tariff Schedule of the United States (2004) -- Supplement 1 Annotated for Statistical Reporting Purposes
Harmonized Tariff Schedule of the United States (2004) -- Supplement 1 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2004) -- Supplement 1 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABACAVIR 136470-78-5 ACEXAMIC ACID 57-08-9 ABAFUNGIN 129639-79-8 ACICLOVIR 59277-89-3 ABAMECTIN 65195-55-3 ACIFRAN 72420-38-3 ABANOQUIL 90402-40-7 ACIPIMOX 51037-30-0 ABARELIX 183552-38-7 ACITAZANOLAST 114607-46-4 ABCIXIMAB 143653-53-6 ACITEMATE 101197-99-3 ABECARNIL 111841-85-1 ACITRETIN 55079-83-9 ABIRATERONE 154229-19-3 ACIVICIN 42228-92-2 ABITESARTAN 137882-98-5 ACLANTATE 39633-62-0 ABLUKAST 96566-25-5 ACLARUBICIN 57576-44-0 ABUNIDAZOLE 91017-58-2 ACLATONIUM NAPADISILATE 55077-30-0 ACADESINE 2627-69-2 ACODAZOLE 79152-85-5 ACAMPROSATE 77337-76-9 ACONIAZIDE 13410-86-1 ACAPRAZINE 55485-20-6 ACOXATRINE 748-44-7 ACARBOSE 56180-94-0 ACREOZAST 123548-56-1 ACEBROCHOL 514-50-1 ACRIDOREX 47487-22-9 ACEBURIC ACID 26976-72-7