Electrolytic Manganese from Aqueous Sodium Manganate Solution William F
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Precursors and Chemicals Frequently Used in the Illicit Manufacture of Narcotic Drugs and Psychotropic Substances 2017
INTERNATIONAL NARCOTICS CONTROL BOARD Precursors and chemicals frequently used in the illicit manufacture of narcotic drugs and psychotropic substances 2017 EMBARGO Observe release date: Not to be published or broadcast before Thursday, 1 March 2018, at 1100 hours (CET) UNITED NATIONS CAUTION Reports published by the International Narcotics Control Board in 2017 The Report of the International Narcotics Control Board for 2017 (E/INCB/2017/1) is supplemented by the following reports: Narcotic Drugs: Estimated World Requirements for 2018—Statistics for 2016 (E/INCB/2017/2) Psychotropic Substances: Statistics for 2016—Assessments of Annual Medical and Scientific Requirements for Substances in Schedules II, III and IV of the Convention on Psychotropic Substances of 1971 (E/INCB/2017/3) Precursors and Chemicals Frequently Used in the Illicit Manufacture of Narcotic Drugs and Psychotropic Substances: Report of the International Narcotics Control Board for 2017 on the Implementation of Article 12 of the United Nations Convention against Illicit Traffic in Narcotic Drugs and Psychotropic Substances of 1988 (E/INCB/2017/4) The updated lists of substances under international control, comprising narcotic drugs, psychotropic substances and substances frequently used in the illicit manufacture of narcotic drugs and psychotropic substances, are contained in the latest editions of the annexes to the statistical forms (“Yellow List”, “Green List” and “Red List”), which are also issued by the Board. Contacting the International Narcotics Control Board The secretariat of the Board may be reached at the following address: Vienna International Centre Room E-1339 P.O. Box 500 1400 Vienna Austria In addition, the following may be used to contact the secretariat: Telephone: (+43-1) 26060 Fax: (+43-1) 26060-5867 or 26060-5868 Email: [email protected] The text of the present report is also available on the website of the Board (www.incb.org). -

MATERIAL SAFETY DATA SHEET Sodium Permanganate Solution
MATERIAL SAFETY DATA SHEET Sodium Permanganate solution Page 1 to 4 1: Company and Product Identification Chemical Product Sodium Permanganate Trade Name Sodium Permanganate 20% Sodium Permanganate 40% Synonym NaMnO4 Permanganic acid of Sodium Salt Company Name Magnesia Chemicals LLP Address 81/A, Parhar Village road, Lonand, Tal. Phaltan, Dist. Satara, Maharashtra. Pin: 415523. India Telephone +91 7558428995 Fax E mail [email protected] Website www.magnesiachemicals.com 2: Hazards Identification 1. EYE CONTACT - Sodium Permanganate is damaging to eye tissue on contact. It may cause burns that result in damage to the eye. 2. SKIN CONTACT - Momentary contact of solution at room temperature may be irritating to the skin, leaving brown stains. Prolonged contact is damaging to the skin. 3. INHALATION - Acute inhalation toxicity data are not available. However, airborne concentrations of sodium permanganate in the form of mist may cause irritation to the respiratory tract. 4. INGESTION - Sodium permanganate solution, if swallowed, may cause burns to mucous membranes of the mouth, throat, oesophagus, and stomach. 3: Hazard Description &Symbols MATERIAL CAS NO. EINECS % HAZARD DATA Sodium Permanganate 10101-50-5 233-251-1 20-40 PEL/C 5 mg Mn per cubic meter of air TLV-TWA 0.2 mg Mn per cubic meter of air SYMBOLS: Oxidizing Irritant Environmental Chemical Hazard RISK PHRASES: 8 - Contact with combustibles may case fire. 22 - Harmful if swallowed. 50/53 - Very toxic to aquatic organisms, may cause long-term effects in the aquatic environment. SAFETY PHRASES: 17 - Keep away from combustible materials. 24/25 - Avoid contact with skin and eyes. 26 - In case of contact with eyes, rinse immediately with plenty of water and seek medical advice. -

Carbonyl Compounds
Carbonyl Compounds What are Carbonyl Compounds? Carbonyl compounds are compounds that contain the C=O (carbonyl) group. Preparation of Aldehydes: 1. Preparation from Acid Chloride (Rosenmund Reduction): This reaction was named after Karl Wilhelm Rosenmund, who first reported it in 1918. The reaction is a hydrogenation process in which an acyl chloride is selectively reduced to an aldehyde. The reaction, a hydrogenolysis, is catalysed by palladium on barium sulfate, which is sometimes called the Rosenmund catalyst. 2. Preparation from Nitriles: This reaction involves the preparation of aldehydes (R-CHO) from nitriles (R- CN) using SnCl2 and HCl and quenching the resulting iminium salt ([R- + − CH=NH2] Cl ) with water (H2O). During the synthesis, ammonium chloride is also produced. The reaction is known as Stephen Aldehyde synthesis. Dr. Sumi Ganguly Page 1 3. Preparation from Grignard Reagent: When Grignard Reagent is reacted with HCN followed by hydrolysis aldehyde is produced. Preparation of Ketones: 1. Preparation from Acid Chloride (Friedel-Crafts Acylation): Acid chlorides when reacted with benzene in presence of anhydrous AlCl3, aromatic ketone are produced. However, only aromatic ketones can be prepared by following this method. In order to prepare both aromatic and aliphatic ketones acid chlorides is reacted with lithium dialkylcuprate (Gilman Reagnt). Dr. Sumi Ganguly Page 2 The lithium dialkyl cuprate is produced by the reaction of two equivalents of the organolithium reagent with copper (I) iodide. Example: 3. Preparation from Nitriles and Grignard Reagents: When Grignard Reagent is reacted with RCN followed by hydrolysis aldehyde is produced. Dr. Sumi Ganguly Page 3 Physical Characteristic of Carbonyl Compounds: 1) The boiling point of carbonyl compounds is higher than the alkanes with similar Mr. -

Manufacturing of Potassium Permanganate Kmno4 This Is the Most Important and Well Known Salt of Permanganic Acid
Manufacturing of Potassium Permanganate KMnO4 This is the most important and well known salt of permanganic acid. It is prepared from the pyrolusite ore. It is prepared by fusing pyrolusite ore either with KOH or K2CO3 in presence of atmospheric oxygen or any other oxidising agent such as KNO3. The mass turns green with the formation of potassium manganate, K2MnO4. 2MnO2 + 4KOH + O2 →2K2MnO4 + 2H2O 2MnO2 + 2K2CO3 + O2 →2K2MnO4 + 2CO2 The fused mass is extracted with water. The solution is now treated with a current of chlorine or ozone or carbon dioxide to convert manganate into permanganate. 2K2MnO4 + Cl2 → 2KMnO4 + 2KCl 2K2MnO4 + H2O + O3 → 2KMnO4 + 2KOH + O2 3K2MnO4 + 2CO2 → 2KMnO4 + MnO2 + 2K2CO3 Now-a-days, the conversion is done electrolytically. It is electrolysed between iron cathode and nickel anode. Dilute alkali solution is taken in the cathodic compartment and potassium manganate solution is taken in the anodic compartment. Both the compartments are separated by a diaphragm. On passing current, the oxygen evolved at anode oxidises manganate into permanganate. At anode: 2K2MnO4 + H2O + O → 2KMnO4 + 2KOH 2- - - MnO4 → MnO4 + e + - At cathode: 2H + 2e → H2 Properties: It is purple coloured crystalline compound. It is fairly soluble in water. When heated alone or with an alkali, it decomposes evolving oxygen. 2KMnO4 → K2MnO4 + MnO2 + O2 4KMnO4 + 4KOH → 4K2MnO4 + 2H2O + O2 On treatment with conc. H2SO4, it forms manganese heptoxide via permanganyl sulphate which decomposes explosively on heating. 2KMnO4+3H2SO4 → 2KHSO4 + (MnO3)2SO4 + 2H2O (MnO3)2SO4 + H2O → Mn2O7 + H2SO4 Mn2O7 → 2MnO2 + 3/2O2 Potassium permanganate is a powerful oxidising agent. A mixture of sulphur, charcoal and KMnO4 forms an explosive powder. -

Revision Guide
Revision Guide Chemistry - Unit 3 Physical and Inorganic Chemistry GCE A Level WJEC These notes have been authored by experienced teachers and are provided as support to students revising for their GCE A level exams. Though the resources are comprehensive, they may not cover every aspect of the specification and do not represent the depth of knowledge required for each unit of work. 1 Content Page Section 2 3.1 – Redox and standard electrode potential 13 3.2 - Redox reactions 20 3.3 - Chemistry of the p-block 30 3.4 - Chemistry of the d-block transition metals 35 3.5 - Chemical kinetics 44 3.6 - Enthalpy changes for solids and solutions 50 3.7 - Entropy and feasibility of reactions 53 3.8 - Equilibrium constants 57 3.9 - Acid-base equilibria 66 Acknowledgements 2 3.1 – Redox and standard electrode potential Redox reactions In AS, we saw that in redox reactions, something is oxidised and something else is reduced (remember OILRIG – this deals with loss and gain of electrons). Another way that we can determine if a redox reaction has happened is by using oxidation states or numbers (see AS revision guide pages 2 and 44). You need to know that: - • oxidation is loss of electrons • reduction is gain of electrons • an oxidising agent is a species that accepts electrons, thereby helping oxidation. It becomes reduced itself in the process. • a reducing agent is a species that donates electrons, thereby helping reduction. It becomes oxidised itself in the process. You also should remember these rules for assigning oxidation numbers in a compound: - 1 All elements have an oxidation number of zero (including diatomic molecules like H2) 2 Hydrogen is 1 unless it’s with a Group 1 metal, then it’s -1 3 Oxygen is -2 (unless it’s a peroxide when it’s -1, or reacted with fluorine, when it’s +2). -

DEA Regulated Chemical Initiatives
Christine A. Sannerud, Chief Drug and Chemical Evaluation Section (ODE) U.S. Drug Enforcement Administration (202) 307-7183 Office of Diversion Control, ODE 1 Drug and Chemical Evaluation Section (ODE) Chemical Role includes: • Determination of chemicals used in the manufacture of controlled substances. • Use of existing regulatory mechanisms to control compounds used in this manufacture. Office of Diversion Control, ODE 2 Current Activities • Iodine Regulations (NPRM) • Sodium Permanganate • Elimination of Ephedrine/Pseudoephedrine Chemical Mixture Exemptions • Control of Precursors to Fentanyl • Positional Isomers Definition Office of Diversion Control, ODE 3 Methamphetamine Production Office of Diversion Control, ODE 4 Iodine Control • Iodine used as reagent in Methamphetamine production • Number one U.S. clandestinely produced drug Office of Diversion Control, ODE 5 U.S. Department of Justice Drug Enforcement Administration Office of Diversion Control Chemical Investigations Section Methamphetamine Clandestine Laboratory Seizures With DEA participation WASH MONTANA N DAKOTA WASH MONTANA N DAKOTA WASH MONTANA N DAKOTA MN 1993 -218 MN 1994 -263 MN 1995 -327 ME ME WI ME S DAKOTA WI S DAKOTA WI OREGON S DAKOTA OREGON VT OREGON VT 10 VT WYOMING IDAHO WYOMING WYOMING MI IDAHO MI MI IDAHO NH NH NH IOWA NY NY NY MA IOWA MA IOWA MA NEBRASKA ILL NEBRASKA ILL NEBRASKA ILL NEVADA NEVADA IN NEVADA IN PA RI OHIO PA RI OHIO PA RI IN OHIO CT CT CT UTAH UTAH COLORADO UTAH COLORADO NJ 14 NJ KANSAS NJ CA 10 COLORADO W CA W CA W KANSAS V DE 25 KANSAS -

Sodium Permanganate Etch Baths and Their Use in Cleaning, Desmearing And/Or Etching Resinous Substrates
Europaisches Patentamt J European Patent Office Publication number: 0 207 586 Office europeen des brevets B1 EUROPEAN PATENT SPECIFICATION Date of publication of patent specification: 22.11.90 int. ci.5: C 23 C 18/22, H 05 K 3/42, C 09 K 13/02 Application number: 86302336.2 Date of filing: 27.03.86 Sodium permanganate etch baths and their use in cleaning, desmearing and/or etching resinous substrates. (§) Priority: 31.05.85 US 739726 Proprietor: MORTON INTERNATIONAL, INC. 110 North Wacker Drive Chicago Illinois 60606 (US) Date of publication of application: 07.01.87 Bulletin 87/02 Inventor: Krulik, Gerald 25142 Barents Street Publication of the grant of the patent- Laguna Hills California 92653 (US) 22.11.90 Bulletin 90/47 Representative: Bankes, Stephen Charles Digby Designated Contracting States- etal AT BE CH DE FR GB IT LI LU NL SE BARON & WARREN 18 South End Kensington London W8 5BU (GB) 1 References cited: US-A-3080323 US-A-3 095 380 US-A-3 528924 US-A-3 652 351 US-A-3 652 417 <0 00 IO N o CM o Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. Notice of opposition shall Q. be filed in a written reasoned statement. It shall not be deemed to have been filed until the opposition fee has been Ill paid. (Art. 99(1 ) European patent convention). Courier Press, Leamington Spa, England. -

Jnited States Patent Office Patented July 9, 1957
2,798,878 Jnited States Patent Office Patented July 9, 1957 2 weeks; and, finally, the method must be safe from the hazard of-explosion. One of the objects of the invention is to provide a 2,798,878 method of oxidizing graphite to graphitic oxide with the PREPARATION OF GRAPHITIC ACID production of a high quality product of low carbon-to Williaria S. Hummers, Jr., Detroit, Mich., assignor to Na oxygen ratio. tional Lead Company, New York, N. Y., a corporation Another object of the invention is to provide a method of New Jersey of preparing graphitic oxide in a hazard-free manner with a processing time of one or two hours, or less. No Drawing. Application July 19, 1954, 0. Another object of the invention is to provide a method Seria No. 444,400 of preparing graphitic oxide in high yield, with a favor ably low consumption of oxidizing agents. 16 Claims. (C. 260-348) Another object of the invention is to provide a method of preparing graphitic oxide with materials which are This invention relates to the preparation of graphitic 5 relatively cheap, available, and readily handled. oxide from graphite, and more particularly is concerned Other objects of the invention will appear as the de with an improved method of carrying out the oxidation scription thereof proceeds. In accordance with the invention, the starting material, step involved. which is a graphitic substance which may be graphite, Graphitic oxide, sometimes known as graphitic acid, 20 has been known for almost a century, and is an important and is preferably a high grade of synthetic or natural material for experimental studies in electrode processes graphite, such as Ceylon graphite, is treated with a water of colloidal properties of materials related to humic acids, free mixture of concentrated sulphuric acid and an an in inorganic structural chemistry, and the like. -
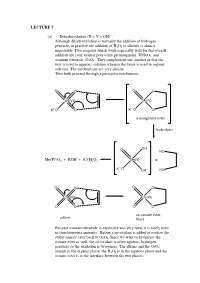
A Manganate Ester Hydrolysis Mn(IV)
LECTURE 7 (a) Dihydroxylation (X = Y = OH) Although dihydroxylation is formally the addition of hydrogen peroxide, in practice the addition of H2O2 to alkenes is almost impossible. Two reagents which work especially well for this overall addition are cold, neutral potassium permanganate, KMnO4, and osmium tetroxide, OsO4. They complement one another in that the first is used in aqueous solution whereas the latter is used in organic solvents. The mechanisms are very similar. Thus both proceed through a pericyclic mechanism: O O O O Mn(VII) Mn(V) +- K+-O O K O O a manganate ester hydrolysis O OH HO Mn(IV)O2 + KOH + 0.5 H2O2 Mn(V) + +- K O OH HO O O O O Os(VIII) Os(VI) O O O O an osmate ester, yellow black Because osmium tetroxide is expensive and very toxic it is rarely used in stoichiometric amounts. Rather a co-oxidant is added to oxidise the stable osmate ester back to OsO4. Since we want to hydrolyse the osmate ester as well, the co-oxidant is often aqueous hydrogen peroxide so the oxidation is two-phase. The alkene and the OsO4 remain in the organic phase, the H2O2 is in the aqueous phase and the osmate ester is at the interface between the two phases: H OH H OH H OsO4, + aq.H2O2, CH Cl H 2 2 OH H H OH (c) Epoxidation (X = Y = O) The reaction of alkenes with peroxy acids (RCO3H) leads to a cyclic ether, known as an epoxide, as follows: R R O O O H O H O O Although the reaction will work with peroxyacetic acid (R = Me) it works best with peroxy acids bearing electron-withdrawing groups e.g. -

The True Formula
ON THE TRUE FORMULA FOR PERMANGANATES. DISSERTATION PRESENTED TO THE FACULTY OF THE UNIVERSITT7 ' OF VIRGINIA. In application fir the Degree of Dadar of 'Pflilawplzj'. BY CHARLES M. B‘RADBURY, 0F VIRGINIA. THESIS —Permanga;zz}: acid is moiwéaszc, and the per-:V _ mangamztes are represented 6}! the generalfbmwla R (Mn 04 p. -' : . ; V To His Fat/tor, 31m ‘18. granary, (35511. 77:13 Paper is Inscribed IN TOKEN OF AFFECTION, BY film 9mm, Preface. The greater part of the following paper was written nearly a year ago. As it was nearing completion, I found, from an allusion in .an article by M. Raoult, of Paris, (see Bzzlleiz'n dc Soc. C/u'm. de Paris, XLVI, p. 805), that my conclusion as to the true formula for permanganates had been reached already by that eminent chemist, by means of a new method, origi- nated by himself, for determining molecular weights. Upon examining M. Raoult‘s original papem‘fHéWIz—Iifilc C/zz'm. ct dc Ply/5., (6), VIII, July, 1886, p. 3 30, however, it was found that nothing was given concerning permanganates, beyond the simple statement that they were shown by the method to be monobasic. T Dr. Victor Meyer and Dr. Auwers, of Gottingen, have recently extended and simplified the method of M. Raoult, (see Berk/zit a’n’ Dads. Chem. 656115., for February 27 and March 12, 1888), and I am therefore enabled to add materially to the evidence already collected in the dissertation, by a con- vincing application of this method. ‘ Had the detailed results of M. -

Interagency Committee on Chemical Management
DECEMBER 15, 2020 INTERAGENCY COMMITTEE ON CHEMICAL MANAGEMENT EXECUTIVE ORDER NO. 02-19 REPORT TO THE GOVERNOR WALKE, PETER Table of Contents Executive Summary ...................................................................................................................... 2 I. Introduction ............................................................................................................................. 4 II. Chemical Nomination Review Framework .......................................................................... 6 III. Summary of Chemical Use in the State Based on Reported Chemical Inventories.......... 8 IV. Summary of Identified Risks to Human Health and the Environment from Reported Chemical Inventories .............................................................................................................. 9 V. Summary of any change under Federal Statute or Rule affecting the Regulation of Chemicals in the State ............................................................................................................ 9 VI. Recommended Legislative or Regulatory Action to Reduce Risks to Human Health and the Environment from Regulated and Unregulated Chemicals of Emerging Concern . 25 VII. Final Thoughts ................................................................................................................. 26 Appendices ................................................................................................................................... 27 1 Executive Summary On August 7, 2017, Governor -

Interagency Committee on Chemical Management
DECEMBER 14, 2018 INTERAGENCY COMMITTEE ON CHEMICAL MANAGEMENT EXECUTIVE ORDER NO. 13-17 REPORT TO THE GOVERNOR WALKE, PETER Table of Contents Executive Summary ...................................................................................................................... 2 I. Introduction .......................................................................................................................... 3 II. Recommended Statutory Amendments or Regulatory Changes to Existing Recordkeeping and Reporting Requirements that are Required to Facilitate Assessment of Risks to Human Health and the Environment Posed by Chemical Use in the State ............................................................................................................................ 5 III. Summary of Chemical Use in the State Based on Reported Chemical Inventories....... 8 IV. Summary of Identified Risks to Human Health and the Environment from Reported Chemical Inventories ........................................................................................................... 9 V. Summary of any change under Federal Statute or Rule affecting the Regulation of Chemicals in the State ....................................................................................................... 12 VI. Recommended Legislative or Regulatory Action to Reduce Risks to Human Health and the Environment from Regulated and Unregulated Chemicals of Emerging Concern ..............................................................................................................................