Some Properties of Platinum Pentafluoride and Some
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

2018 Annual Survey of Biological and Chemical Agents Regulated by Homeland Security (And Carcinogens Regulated by OSHA)
Name: Dept: Date: 2018 Annual Survey of Biological and Chemical Agents regulated by Homeland Security (and carcinogens regulated by OSHA) Due (date) All labs that do not have a current chemical inventory in Chematix MUST complete this survey. The University is required to make an annual report of all chemicals on the Chemical Facility Anti-Terrorism Standards (CFATS) lists. Additional information regarding the regulations is available on the EH&S website at http://www.safety.rochester.edu/restricted/occsafe/chemicalagent.html and https://www.selectagents.gov. 1. Please review the lists on the following pages and indicate if any are possessed by your lab. The CAS# has been added to the list for ease of searching databases. The CAS# is a Chemical Abstract Service numbering system which assigns a unique number to every chemical substance based on structure; this helps avoid confusion by use of synonyms or different naming conventions. a. If yes for possession, place an X in the applicable box and if requested, include the quantity held in your lab. b. If no, leave blank. 2. After reviewing the list, please complete the information box below (or on last page for possession), then sign, date and return to EH&S. 3. Please call Donna Douglass at 275-2402 if you have any questions. Thank you for your cooperation in collecting data required by the Department of Homeland Security! Possession: 1) Fill in applicable boxes, 2) have PI sign last page, 3) return all pages to Donna Douglass OR Non-possession: 1) Check only one box on the left, 2) sign, 3) return just this page to Donna Douglass I do not have a lab, do not work in a lab, nor do I possess any of the agents in this survey. -

The Oxidizing Behavior of Some Platinum Metal Fluorides
Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory Title THE OXIDIZING BEHAVIOR OF SOME PLATINUM METAL FLUORIDES Permalink https://escholarship.org/uc/item/46b5r4v5 Author Graham, Lionell Publication Date 1978-10-01 eScholarship.org Powered by the California Digital Library University of California LBL-8088.c,y '': ·,~, . ' '~~.. .· J!' ... THE OXIDIZING BEHAV!OROF SOME PLATINUM METAL FLUORIDES . Li onell Graham. · (Ph~ D. Thesis ) IIi !l1/ "~ · r-,> ·1cr7o October 1978 ~t .. ~, ~L l ... ;,() · .. t .. J r.~~ r: !\ ~;~, ·l .• ~~:J"~~ t~J ;,~.)(·_ .. {·~ tJ 1v~ ;;:: r~- J··r·r,;~ Ei:~-:·~,:~~ <'11 ().i\{ Prep~red for the .. U. S. Department of Energy. under Contract W~7405~ENG-48 TWO-WEEK LOAN COPY This is a Library Circulating Copy which may be borrowed for two weeks. .. ' ·t•·! •. ·. For a personal retention copy, call Tech. Info. Diu is ion, Ext. 6782 .. LEGAL NOTICE -___,..-----~__, This report was prepared as an account of work sponsored by the United States Government Neither the United States nor the Depart ment of Energy, nor any of their employees, nor any of their con tractors, subcontractors, or their employees, makes any warraf')ty, express or implied,orassumes any legal liabii bility for the accuracy, completeness or usefulness of nformation, appa- ratus, product. ·· · · disclosed; or rep ·. ts that its use would. not infringe p. · · owned rights ... -i- THE OXIDIZING BEHAVIOR OF SOME PLATINUM METAL FLUORIDES Contents Abstract . v I. General Introduction 1 II. General Apparatus and Handling Techniques 2 A. Apparatus . 2 1. General 2 2. The Cryogenic Technology, Inc., Model 21 Cryo-Cooler .. 3 3. Reactor for Gas-Gas Reaction Products 5 B. -
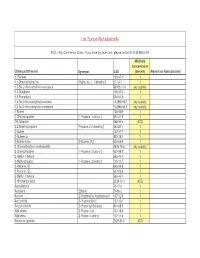
Homeland Security List
List Sorted Alphabetically ACG = Any Commercial Grade. If you have any questions, please contact EHS @ 898-5126. Minimum Concentration Chemical Of Interest Synonym CAS (percent) Amount on Hand (pounds) 1- Pentene 109-67-1 1 1,1-Dimethylhydrazine [Hydrazine, 1, 1-dimethyl-] 57-14-7 1 1,3-Bis(2-chloroethylthio)-n-propane 63905-10-2 any quantity 1,3-Butadiene 106-99-0 1 1,3-Pentadiene 504-60-9 1 1,4-Bis(2-chloroethylthio)-n-butane 142868-93-7 any quantity 1,5-Bis(2-chloroethylthio)-n-pentane 142868-94-8 any quantity 1-Butene 106-98-9 1 1-Chloropropylene [1-Propene, 1-chloro-] 590-21-6 1 1H-Tetrazole 288-94-8 ACG 2,2-Dimethylpropane [Propane, 2,2-dimethyl-] 463-82-1 1 2-Butene 107-01-7 1 2-Butene-cis 590-18-1 1 2-Butene-trans [2-Butene, (E)] 624-64-6 1 2-Chloroethylchloro-methylsulfide 2625-76-5 any quantity 2-Chloropropylene [1-Propene, 2-chloro-] 557-98-2 1 2-Methyl-1-butene 563-46-2 1 2-Methylpropene [1-Propene, 2-methyl-] 115-11-7 1 2-Pentene, (E)- 646-04-8 1 2-Pentene, (Z)- 627-20-3 1 3-Methyl-1-butene 563-45-1 1 5-Nitrobenzotriazol 2338-12-7 ACG Acetaldehyde 75-07-0 1 Acetylene [Ethyne] 74-86-2 1 Acrolein [2-Propenal] or Acrylaldehyde 107-02-8 1 Acrylonitrile [2-Propenenitrile] 107-13-1 1 Acrylyl chloride [2-Propenoyl chloride] 814-68-6 1 Allyl alcohol [2-Propen-1-ol] 107-18-6 1 Allylamine [2-Propen-1-amine] 107-11-9 1 Aluminum (powder) 7429-90-5 ACG Ammonia (anhydrous) 7664-41-7 1 Ammonia (conc. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Qt5km53474.Pdf
Lawrence Berkeley National Laboratory Recent Work Title REDOX REACTIONS IN THE XeF2/PLATINUM FLUORIDE and XeF2/PALLADIUM FLUORIDE SYSTEMS and THE CONVERSION OF XeF2 T0 XeF4 and Xe Permalink https://escholarship.org/uc/item/5km53474 Author Bartlett, Neil Publication Date 1975-10-01 eScholarship.org Powered by the California Digital Library University of California Submitted to Journal of Fluorine LBL-4195 Chemistry Preprint c.\ REDOX REACTIONS IN THE XeF 2/PLA TINUM FLUORIDE AND XeFz/ PALLADIUM FLUORIDE SYSTEMS AND THE CONVERSION OF XeF 2 TO XeF 4 AND Xe Neil Bartlett, Boris Zemva and Lionell Graham October 1975 ) Prepared for the U. S. Energy Research and Development Administration under Contract W -7405-ENG-48 · For Reference Not to be taken from this room t"' tJ:j t"' I I"\ ,j:>. ....... -..D -- (J'1 r- r r·· r r. n DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. -

CSAT Top-Screen Questions OMB PRA # 1670-0007 Expires: 5/31/2011
CSAT Top-Screen Questions January 2009 Version 2.8 CSAT Top-Screen Questions OMB PRA # 1670-0007 Expires: 5/31/2011 Change Log .........................................................................................................3 CVI Authorizing Statements...............................................................................4 General ................................................................................................................6 Facility Description.................................................................................................................... 7 Facility Regulatory Mandates ................................................................................................... 7 EPA RMP Facility Identifier....................................................................................................... 9 Refinery Capacity....................................................................................................................... 9 Refinery Market Share ............................................................................................................. 10 Airport Fuels Supplier ............................................................................................................. 11 Military Installation Supplier................................................................................................... 11 Liquefied Natural Gas (LNG) Capacity................................................................................... 12 Liquefied Natural Gas Exclusion -

· for Reference
Submitted to Journal of Fluorine LBL-4195 Chemistry Preprint c.\ REDOX REACTIONS IN THE XeF 2/PLA TINUM FLUORIDE AND XeFz/ PALLADIUM FLUORIDE SYSTEMS AND THE CONVERSION OF XeF 2 TO XeF 4 AND Xe Neil Bartlett, Boris Zemva and Lionell Graham October 1975 ) Prepared for the U. S. Energy Research and Development Administration under Contract W -7405-ENG-48 · For Reference Not to be taken from this room t"' tJ:j t"' I I"\ ,j:>. ....... -..D -- (J'1 r- r r·· r r. n DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof or the Regents of the University of California. -1- REDOX REACTIONS IN THE XeFz/PLATINUM FLUORIDE AND XeFz/PALLADIID-1 FLUORIDE SYSTEMS AND THE CONVERSION OF XeFz TO XeF4 AND Xe \J NEIL BARTLETT, BORIS ZEMVA AND LIONELL GRAHAJ1 Department of Chemistry, University of California, and Inorganic Haterials Research Division, Lawrence Berkeley Laboratory, Berkeley, California 94720 (U.S.A.) (To Professor George H. -

Liquid Oxygen of Argon
USOO6780390B2 (12) United States Patent (10) Patent No.: US 6,780,390 B2 Bhagat et al. (45) Date of Patent: Aug. 24, 2004 (54) METHOD OF PRODUCING HIGH PURITY OTHER PUBLICATIONS GERMANIUM TETRAFLUORIDE Dennis et al., “Germanium. XXI Germanium Tetrafluoride', (75) Inventors: Sudhir Solomon Bhagat, Tulsa, OK Z. Phys. Chem. 130 (1927), pp. 520–531.* (US); Dayal T. Meshri, Tulsa, OK Adams, et al. “The Preparation and Some Properties of New (US); Sanjay D. Meshri, Tulsa, OK Germanium Flouride”. J. Inorg. Nucl. Chem., 1971, vol. 33, (US); Michale Shane Petty, Claremore, pp. 1301-1306, no month. OK (US) Burg, Anton B. “Germanium Tetrafluoride”, Fluorine Chem istry, Edited by Dr. J.H. Simons. Academic Press Inc., (73) Assignee: Advance Research Chemicals, Inc., Publishers, New York, NY. 1950. vol. 1, pp. 110-111, Catoosa, OK (US) 118-119, no month. Hiroyuki et al. “Production of Germanium Tetrafluoride (*) Notice: Subject to any disclaimer, the term of this Having High Purity”. PAJ 00–23–76, 01051329 JP, NDN patent is extended or adjusted under 35 190-0003-9421-0, no month. U.S.C. 154(b) by 173 days. Isao et al. "Production of Germanium Tetrafluoride'. PAJ 08-01-94, 06234523 JP, NDN- 190-0171-8558-1. (21) Appl. No.: 10/218,894 * cited by examiner (22) Filed: Aug. 14, 2002 Primary Examiner Ngoc-Yen Nguyen Prior Publication Data (74) Attorney, Agent, or Firm-Fellers, Snider, (65) Blankenship, Bailey & Tippens, P.C. US 2004/0076577 A1 Apr. 22, 2004 (57) ABSTRACT (51) Int. Cl." ............................ C01G 17/04; CO1B 9/08 (52) U.S. Cl. -

New Regs Announced for Securing High-Risk
Number 586 April 9, 2007 Client Alert Latham & Watkins Environment, Land & Resources Department Department of Homeland Security Announces New Regulations for Securing High-Risk Chemical Facilities Summary effective 60 days after publication in the Federal Register, the Department The Department of Homeland Security has sought additional comment within plans to impose new requirements on 30 days on one key component of the chemical facilities that possess listed regulations. chemicals above specified quantities, and in many cases will require facilities What Facilities Are Affected? to prepare vulnerability assessments, develop security plans, and implement Under these regulations, the Department and install a suite of security measures. will screen tens of thousands of These requirements could begin to take “chemical facilities” for vulnerability We recommend effect for certain facilities as early as ” to and potential consequences of a June 2007. that all companies terrorist attack, and will regulate that possess or those it determines to be “high-risk.” otherwise handle Background “Chemical facility” is defined broadly as “any establishment that possesses or chemicals review In the four and a half years since the plans to possess, at any point in time, the regulations.“ terrorist attacks of September 11, 2001, a quantity of a chemical substance Congress has debated, but until recently determined by the Secretary to be failed to pass, legislation regulating the potentially dangerous or that meets security of chemical facilities in the US. other risk-related criteria identified On October 4, 2006, the Department by the Department.” A “high-risk” of Homeland Security Appropriations facility is one which, “in the discretion Act of 2007 (the Act) was signed into of the Secretary of Homeland Security, law. -

THE FLUORIDES of PLATINUM and R E L a T E D Compounds by DEREK HARRY LOHMANN B.Sc, University of London, 1953 M.Sc, Queen's Univ
THE FLUORIDES OF PLATINUM and related compounds by DEREK HARRY LOHMANN B.Sc, University of London, 1953 M.Sc, Queen's University, Ontario, 1959 A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in the Department of CHEMISTRY We accept this thesis as conforming to the required standard. THE UNIVERSITY OF BRITISH COLUMBIA October 1961, In presenting this thesis in partial fulfilment of the requirements for an advanced degree at the University of British Columbia, I agree that the Library shall make it freely available for reference and study. I further agree that- permission for extensive copying of this thesis for scholarly purposes may be granted by the Head of my Department or by his representatives. It is understood that copying or publication of this thesis for financial gain shall not be allowed without my written permission. Department of CHEMISTRY The University of British Columbia, Vancouver 8, Canada. Date 31st October 1961. %\\t Pntesttg of ^irtttslj Columbia FACULTY OF GRADUATE STUDIES PROGRAMME OF THE FINAL ORAL EXAMINATION FOR THE DEGREE OF DOCTOR OF PHILOSOPHY PUBLICATIONS oi . I. "Polar effects in Hydrogen abstraction reactions," M. P. Godsay, DEREK HARRY LOHMANN D. H. Lohmann and K. E. Russell, Chem. and Ind., 1959, 1603. B.Sc, University of London, 1953 M.Sc, Queen's University, Ontario, 1959 2. "Two new fluorides of Platinum," N. Bartlett and D. H. Lohmann, Proc. Chem. Soc, 1960, 14. TUESDAY, NOVEMBER 28th, 1961, AT 1:00 P.M. 3. "The reaction of 2,2-Diphenyl-l-Picrylhydrazyl with 9,10-Dihy- droanthracene and 1,4-Dihydronaphthalene," I. -

License Application for International Isotopes, Inc
NRC FORM 313 U.S. NUCLEAR REGULATORY COMMISSION APPROVED BY OMB: NO. 3150-0120 EXPIRES: 1013112005 (4-2004) Estimated burden per response to comply with this mandatory collection request: 7 10CFR 30,32,33, hours. Submittal of the application is necessary to determine that the applicant Is 34,35. 36. 39. and 40 qualified and that adequate procedures exist to protect the public health and safety. Send comments regarding burden estimate to the Records and FOAIAPrivacy Services Brnd chmments(T- US. NuclearRegulatmoaryCommission, Washington, DC 20555-000, or by Internet e-mail to Infocolects@ nrc. go, and to the Desk Officer, Office of InfrmaionandReglatryffarsNEO1 002,(35~0I020), Office of Management APPLICATION FOR MATERIAL LICENSE andaBudget, Washington. DC 20503.lIfa me ans used to impose an Information collection does not display a currently valid OMB control number, the NRC may not conduct or sponsor, and a person is not required to respond to, the Information INSTRUCTIONS: SEE THE APPROPRIATE LICENSE APPLICATION GUIDE FOR DETAILED INSTRUCTIONS FOR COMPLETING APPLICATION. SEND TWO COPIES OF THE ENTIRE COMPLETED APPLICATION TO THE NRC OFFICE SPECIFIED BELOW. APPLICATION FOR DISTRIBUTION OF EXEMPT PRODUCTS FILE APPLICATIONS WITH: IF YOU ARE LOCATED IN: DMSION OF INDUSTRIAL AND MEDICAL NUCLEAR SAFETY IWJNOIS, INDIANA, IOWA, MICHIGAN, MINNESOTA, MISSOURI, OHIO, OR WISCONSIN. SEN) OFFICE OF NUCLEAR MATERIALS SAFETY AND SAFEGUARDS APPLICATIONS TO: U.S. NUCLEAR REGULATORY COMMISSION WASHINGTON. DC 20555-0001 MATERIALS LICENSING BRANCH U.S. NUCLEAR REGULATORY COMMISSION. REGION Ill 2443 WARRENVILLE ROAD. SUITE 210 ALL OTHER PERSONS FILE APPLICATIONS AS FOLLOWS: LISLE, IL 60532R4352 IF YOU ARE LOCATED IN: ALABAMA.