PDF Hosted at the Radboud Repository of the Radboud University Nijmegen
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Stimuli-Responsive Polymersomes and Nanoreactors
Stimuli-responsive polymersomes and nanoreactors Citation for published version (APA): Che, H., & van Hest, J. C. M. (2016). Stimuli-responsive polymersomes and nanoreactors. Journal of Materials Chemistry B, 4(27), 4632-4647 . https://doi.org/10.1039/C6TB01163B DOI: 10.1039/C6TB01163B Document status and date: Published: 17/07/2016 Document Version: Accepted manuscript including changes made at the peer-review stage Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal. -

Curriculum Vitae KRISTI L
Curriculum Vitae KRISTI L. KIICK, PH.D. Professor www.udel.edu/mseg Department of Materials Science and Engineering [email protected] Biomedical Engineering 302 831 0201 University of Delaware, Newark, DE 19716 EDUCATION Doctor of Philosophy Polymer Science and Engineering 2001 University of Massachusetts Amherst, Amherst, MA NDSEG Predoctoral Fellow Master of Science Polymer Science and Engineering 1998 University of Massachusetts Amherst , Amherst, MA Master of Science Chemistry 1991 University of Georgia, Athens, GA NSF Predoctoral Fellow Bachelor of Science Chemistry, Summa cum laude 1989 University of Delaware, Newark, DE Eugene duPont Memorial Scholar RESEARCH INTERESTS Biologically derived methods for the synthesis and assembly of advanced macromolecular materials The development of protein engineering methods for the synthesis of artificial protein polymers functionalized with both natural and non-natural amino acids, and the chemical modification of protein polymers to produce well-defined protein-based architectures. The use of modified proteins in both biological and materials applications such as toxin neutralization, manipulation of cell signaling, and construction of light-emitting devices. The development of new elastomeric materials with well-defined conformational and assembly behavior. The assembly of hydrogel networks via protein-polysaccharide interactions to produce materials that mimic the biological activity of the extracellular matrix and that have controlled mechanical, erosion, and drug delivery properties. The -

Construction and Characterization of Hybrid
CONSTRUCTION AND CHARACTERIZATION OF HYBRID NANOPARTICLES VIA BLOCK COPOLYMER BLENDS AND KINETIC CONTROL OF SOLUTION ASSEMBLY by Yingchao Chen A dissertation submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Material Science and Engineering 2015 Spring © 2015 Yingchao Chen All Rights Reserved ProQuest Number: 3730204 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. ProQuest 3730204 Published by ProQuest LLC (2015). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, MI 48106 - 1346 CONSTRUCTION AND CHARACTERIZATION OF HYBRID NANOPARTICLES VIA BLOCK COPOLYMER BLENDS AND KINETIC CONTROL OF SOLUTION ASSEMBLY by Yingchao Chen Approved: __________________________________________________________ Darrin J. Pochan, Ph.D. Chair of the Department of Material Science and Engineering Approved: __________________________________________________________ Babatunde A. Ogunnaike, Ph.D. Dean of the College of Engineering Approved: __________________________________________________________ James G. Richards, Ph.D. Vice Provost for Graduate and Professional Education I certify that I have read this dissertation and that in my opinion it meets the academic and professional standard required by the University as a dissertation for the degree of Doctor of Philosophy. -
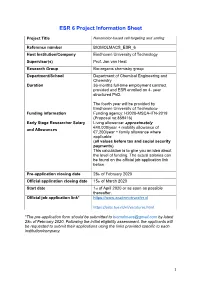
ESR 6 Project Information Sheet
ESR 6 Project Information Sheet Project Title Nanomotor-based cell targeting and sorting Reference number BIOMOLMACS_ESR_6 Host Institution/Company Eindhoven University of Technology Supervisor(s) Prof. Jan van Hest Research Group Bio-organic chemistry group Department/School Department of Chemical Engineering and Chemistry Duration 36-months full-time employment contract provided and ESR enrolled on 4- year structured PhD. The fourth year will be provided by Eindhoven University of Technology Funding information Funding agency: H2020-MSCA-ITN-2019 (Proposal no:859416) Early Stage Researcher Salary Living allowance: approximately €40,000/year + mobility allowance of and Allowances €7,200/year + family allowance where applicable (all values before tax and social security payments) This calculation is to give you an idea about the level of funding. The actual salaries can be found on the official job application link below. Pre-application closing date 28th of February 2020 Official application closing date 15th of March 2020 Start date 1st of April 2020 or as soon as possible thereafter. Official job application link* https://www.academictransfer.nl https://jobs.tue.nl/nl/vacatures.html *The pre-application form should be submitted to [email protected] by latest 28th of February 2020. Following the initial eligibility assessment, the applicants will be requested to submit their applications using the links provided specific to each institution/company. 1 Post Summary Brief description of the project: The main focus of this project will be to develop nanomotors based on polymer vesicles that can recognize, bind and isolate specific cells. Bowl-shaped vesicles, also known as stomatocytes, will be created out of biodegradable block copolymers prepared from poly(ethylene glycol)-poly(D,L- lactic acid) and loaded with enzymes, specifically glucose oxidase and catalase. -

Artificial Cells: Synthetic Compartments with Life-Like Functionality and Adaptivity
This is an open access article published under a Creative Commons Non-Commercial No Derivative Works (CC-BY-NC-ND) Attribution License, which permits copying and redistribution of the article, and creation of adaptations, all for non-commercial purposes. Article pubs.acs.org/accounts Artificial Cells: Synthetic Compartments with Life-like Functionality and Adaptivity Bastiaan C. Buddingh’ and Jan C. M. van Hest* Eindhoven University of Technology, P.O. Box 513 (STO 3.31), 5600 MB Eindhoven, The Netherlands CONSPECTUS: Cells are highly advanced microreactors that form the basis of all life. Their fascinating complexity has inspired scientists to create analogs from synthetic and natural components using a bottom-up approach. The ultimate goal here is to assemble a fully man-made cell that displays functionality and adaptivity as advanced as that found in nature, which will not only provide insight into the fundamental processes in natural cells but also pave the way for new applications of such artificial cells. In this Account, we highlight our recent work and that of others on the construction of artificial cells. First, we will introduce the key features that characterize a living system; next, we will discuss how these have been imitated in artificial cells. First, compartmentalization is crucial to separate the inner chemical milieu from the external environment. Current state-of-the-art artificial cells comprise subcompartments to mimic the hierarchical architecture of eukaryotic cells and tissue. Furthermore, synthetic gene circuits have been used to encode genetic information that creates complex behavior like pulses or feedback. Additionally, artificial cells have to reproduce to maintain a population. -

Shedding the Hydrophilic Mantle of Polymersomes
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/100612 Please be advised that this information was generated on 2021-10-03 and may be subject to change. Polymeric vesicles for drug delivery over the blood-brain barrier and in vivo Imaging René Pascal Brinkhuis Polymeric vesicles for drug delivery over the blood-brain barrier and in vivo imaging Een wetenschappelijke proeve op het gebied van de Natuurwetenschappen, Wiskunde en Informatica Proefschrift ter verkrijging van de graad van doctor aan de Radboud Universiteit Nijmegen op gezag van de rector magnificus prof. mr. S.C.J.J. Kortmann, volgens besluit van het college van decanen in het openbaar te verdedigen op maandag 10 december 2012 om 13.30 uur precies door René Pascal Brinkhuis geboren op 11 april 1981 te Deventer Promotoren: Prof. dr. ir. J.C.M. van Hest Prof. dr. F.P.J.T. Rutjes Manusscriptcommissie: Prof. dr. R.J.M. Nolte Prof. dr. D. Hoekstra (UMC Groningen) Prof. dr. O.C. Boerman (UMC Nijmegen) Druk: Ipskamp drukkers B.V., Enschede ISBN: 978-90-9027254-2 Contents Contents ............................................................................................................................................................................. 3 Preface .............................................................................................................................................................................. -

Meet the Editorial Board DOI: 10.1039/C001033m
PROFILE www.rsc.org/polymers | Polymer Chemistry Meet the Editorial Board DOI: 10.1039/c001033m Professor David Haddleton, for Polymer Research, obtaining her PhD a Member of the Chinese Academy of Editor-in-Chief in 1998 for work in the area of fullerene Sciences. adducts and polymers. A postdoctoral Professor Academician Yang was fellowship with CPIMA (NSF-Center for a faculty member of Fudan University Polymer Interfaces and Macromolecular before he was appointed Vice President Assemblies) brought her to the IBM for Research in 1999. In 2006, he was Almaden Research Center, California appointed Director General of the USA, to work under the direction of Prof. Department of Degree and Postgraduate Craig J. Hawker focusing on the devel- Education, State Council of the People’s opment of new living free polymerization Republic of China. Professor Academi- techniques and approaches to nanoscopic cian Yang serves as senior advisor to the materials. In 2001 she joined XenoPort, Shanghai Municipal Government. He Inc. as a Staff Scientist investigating was inaugurated as President of Fudan enabling technologies for the increased University in January 2009. bioavailability of macromolecular thera- The academic interests of Professor peutics. After this extensive industrial Academician Yang are in condensed experience she started at Vanderbilt matter physics and polymer science. He David Haddleton is Professor of Chem- University as Assistant Professor in the owns many domestic and international istry at the University of Warwick, UK. Department of Chemistry in 2004 with patents and serves on the boards of Professor Haddleton’s research centres a secondary appointment in the Depart- several academic journals including on controlled polymerisation to give ment of Pharmacology, Vanderbilt Chinese Journal of Chemistry and Science macromolecules of designed, desired and Medical School. -

Bioconjugate Chemistry主编 Prof
Chinese Academy of Sciences Key Lab for Biomedical Effects of Nanomaterials and Nanosafety 中科院纳米生物效应与安全性重点实验室 学术报告通知 CAS NS Forum (NO. 328) 演讲者: Prof. Vince Rotello, University of Massachusetts-Amherst, Bioconjugate Chemistry主编 Prof. Jan van Hest, Radboud University Nijmegen (The Netherlands) Bioconjugate Chemistry副主编 Prof. Gang Zheng, University of Toronto, Bioconjugate Chemistry副主编 Prof. Bradley D. Smith, University of Notre Dame, Baltimore County Bioconjugate Chemistry副主编 Prof. Erin Lavik, University of Maryland, Bioconjugate Chemistry副主编 时 间: 2019年10月21日 (星期一) 10:00 地 点: 国家纳米科学中心,南楼二层多功能厅 主持人: 陈春英 研究员 简 介: Vincent Rotello is the Charles A. Goessmann Professor of Chemistry and a University Distinguished Professor at the University of Massachusetts at Amherst. He received his B.S. in Chemistry in 1985 from Illinois Institute of Technology, and his Ph. D. in 1990 in Chemistry from Yale University. He was an NSF postdoctoral fellow at Massachusetts Institute of Technology from 1990-1993, and joined the faculty at the University of Massachusetts in 1993. He has been the recipient of the NSF CAREER and Cottrell Scholar awards, as well as the Camille Dreyfus Teacher- Scholar, the Sloan Fellowships. More recently, he has received the Langmuir Lectureship (2010), and in 2016 he received the Transformational Research and Excellence in Education Award presented by Research Corporation, the Bioorganic Lectureship of the Royal Society of Chemistry (UK), the Australian Nanotechnology Network Traveling Fellowship, and the Chinese Academy of Sciences, President's International Fellowship for Distinguished Researchers. He is a Fellow of both the American Association for the Advancement of Science (AAAS) and of the Royal Society of Chemistry (U.K.). He is also recognized in 2014, 2015 and 2018 by Thomson Reuters/Clarivate as “Highly Cited Researcher”. -

Bioconjugate Chemistry
Editorial pubs.acs.org/bc Editorial t has been an exciting year for us at Bioconjugate Chemistry. show the actual benefits of well-defined bioconjugates to gain I Over the past 12 months we have worked hard to broaden more insight in biological processes and/or to achieve more the scope and impact of the journal while maintaining the rigor efficient therapeutic materials. Besides the translation of that BC was known for. One area we have focused on is existing methods from concept to application, there is also enhancing the manuscript review process. Through meaningful ample room for the design of novel conjugation methodologies. feedback to authors and excellent help from the reviewer A highly important development in the soft matter field is the community, we have lowered the submission to online introduction of stimulus-responsiveness and adaptability in publication to less than 40 days. We have also created a social molecules and materials, to allow them to change their media presencecheck us out at https://www.facebook.com/ properties based on the environment they are in. Bioconjuga- bioconjugatechemistry. Editors and EAB members have tion methods that are responsive and reversible, and which can provided a presence for BC at conferences worldwide, with be executed with spatiotemporal control, will be highly valuable help from the folks at ACS. for the development of this class of bioconjugates, in particular, Coming into Year 2, I thought it would be helpful for you to if this chemistry could be extended to living systems. meet the Editors at BC. This way you can get an idea of what As a polymer chemist by training, I have always been our visions are for the future, and pragmatically whom you’ll interested in approaches that allow the synthesis of highly suggest as Editor for your next submission. -

Biomimicry of Cellular Motility and Communication Based on Synthetic Soft‐Architectures
Biomimicry of Cellular Motility and Communication Based on Synthetic Soft-Architectures Citation for published version (APA): Wang, L., Song, S., van Hest, J., Abdelmohsen, L. K. E. A., Huang, X., & Sánchez, S. (2020). Biomimicry of Cellular Motility and Communication Based on Synthetic Soft-Architectures. Small, 16(27), [1907680]. https://doi.org/10.1002/smll.201907680 Document license: TAVERNE DOI: 10.1002/smll.201907680 Document status and date: Published: 01/07/2020 Document Version: Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers) Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. -

Thesis M. De Ruiter
VIRUS-BASED ORGANELLES ENZYME AND DNA-BASED VIRUS NANOSTRUCTURES AND THEIR CELLULAR INTERACTIONS Mark V. de Ruiter VIRUS-BASED ORGANELLES ENZYME AND DNA-BASED VIRUS NANOSTRUCTURES AND THEIR CELLULAR INTERACTIONS Mark de Ruiter Graduation Committee Chairman: Prof. dr. J. L. Herek University of Twente Promoter: Prof. dr. J. J. L. M. Cornelissen University of Twente Members: Prof. dr. ir. P. Jonkheijm University of Twente Prof. dr. H. B. J. Karperien University of Twente dr. L. I. Segerink University of Twente Prof. dr. C. Wege University of Stuttgart Prof. dr. ir. J. C. M. van Hest Eindhoven University of Technology This work has been funded by the Consolidator Grant (Protcage) of the European Research Council (ERC), “Chemistry in the Confinement of Protein Cages” (project ID 616907). The research in this thesis was conducted within the department of Biomolecular Nanotechnology (BNT), the MESA+ Institute for Nanotechnology and at the faculty of Science and Technology (TNW) of the University of Twente. Copyright ©2019 Mark Vincent de Ruiter, Enschede, The Netherlands PhD Thesis, University of Twente, The Netherlands ISBN: 978-90-365-4703-1 DOI: 10.3990/1.9789036547031 Printed by: Ipskamp printing Cover design by: Mark de Ruiter and Laura de Ruiter - van de Weerd VIRUS-BASED ORGANELLES ENZYME AND DNA-BASED VIRUS NANOSTRUCTURES AND THEIR CELLULAR INTERACTIONS DISSERTATION to obtain the degree of doctor at the University of Twente, on the authority of the rector magnificus, prof. dr. T.T.M. Palstra, on account of the decision of the graduation committee, to be publicly defended on Friday the 1st of February 2019 at 14.45 hours by Mark Vincent de Ruiter born on the 16th of March 1990 in Rhenen, The Netherlands This dissertation has been approved by: Supervisor: Prof. -

16119045.Pdf
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/30138 Please be advised that this information was generated on 2017-12-05 and may be subject to change. ELASTIN AS A BIOMATERIAL FOR TISSUE ENGINEERING Willeke Daamen Cover design and lay-out: Theo Hafmans & Willeke Daamen Elastin as a biomaterial for tissue engineering Daamen, Wilhelmina Francisca Thesis Radboud University Nijmegen Medical Centre, The Netherlands ISBN-10: 9090201947 ISBN-13: 9789090201948 Printed by: Ponsen & Looijen, Wageningen © 2006 by W. Daamen ELASTIN AS A BIOMATERIAL FOR TISSUE ENGINEERING Een wetenschappelijke proeve op het gebied van de Medische Wetenschappen PROEFSCHRIFT ter verkrijging van de graad van doctor aan de Radboud Universiteit Nijmegen op gezag van de Rector Magnifi cus, prof. dr. C.W.P.M. Blom, volgens besluit van het College van Decanen in het openbaar te verdedigen op woensdag 25 januari 2006 des namiddags om 1.30 uur precies door Wilhelmina Francisca Daamen geboren op 19 september 1975 te Druten Promotor: Prof.dr. J.H. Veerkamp Co-promotor: Dr. T.H. van Kuppevelt Manuscriptcommissie: Prof.dr. J.A. Jansen Prof.dr. J. Schalkwijk Prof.dr.ir. J.C.M. van Hest Research presented in this thesis was performed under the supervision of Prof.dr. J.H. Veerkamp and Dr. T.H. van Kuppevelt at the Department of Biochemistry, Nijmegen Centre for Molecular Life Sciences, Radboud University Nijmegen Medical Centre, The Netherlands. The studies were carried out with fi nancial support of Senter as a project of IOP Industrial Proteins (IIE 98012).