Shedding the Hydrophilic Mantle of Polymersomes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Stimuli-Responsive Polymersomes and Nanoreactors
Stimuli-responsive polymersomes and nanoreactors Citation for published version (APA): Che, H., & van Hest, J. C. M. (2016). Stimuli-responsive polymersomes and nanoreactors. Journal of Materials Chemistry B, 4(27), 4632-4647 . https://doi.org/10.1039/C6TB01163B DOI: 10.1039/C6TB01163B Document status and date: Published: 17/07/2016 Document Version: Accepted manuscript including changes made at the peer-review stage Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal. -

Insights Into the Interplay Between Bo3 Oxidase and the Membrane
Constructing artificial respiratory chain in polymer compartments: Insights into the interplay between bo3 oxidase and the membrane Nika Marušicˇa,1, Lado Otrinb,1, Ziliang Zhaoc,1, Rafael B. Lirac,1,2, Fotis L. Kyrilisd,e, ,d,e, Panagiotis L. Kastritisd,e, Tanja Vidakovic-Kochb,3, Ivan Ivanova,3( ﺩﻡﺡﺩﺍﺯﺭﻑ ﯼ) Farzad Hamdi Kai Sundmachera, and Rumiana Dimovac aProcess Systems Engineering, Max Planck Institute for Dynamics of Complex Technical Systems, 39106 Magdeburg, Germany; bElectrochemical Energy Conversion, Max Planck Institute for Dynamics of Complex Technical Systems, 39106 Magdeburg, Germany; cDepartment of Theory and Bio-Systems, Max Planck Institute of Colloids and Interfaces, 14424 Potsdam, Germany; dInterdisciplinary Research Center HALOmem, Martin Luther University Halle-Wittenberg, 06120 Halle/Saale, Germany; and eInstitute of Biochemistry and Biotechnology, Martin Luther University Halle-Wittenberg, 06120 Halle/Saale, Germany Edited by Michael L. Klein, Temple University, Philadelphia, PA, and approved May 14, 2020 (received for review November 5, 2019) Cytochrome bo3 ubiquinol oxidase is a transmembrane protein, the enrichment of the library of building blocks with synthetic which oxidizes ubiquinone and reduces oxygen, while pumping alternatives (in the conventional semantics of chemically syn- protons. Apart from its combination with F1Fo-ATPase to assemble thetic) could improve existing functionalities or introduce new a minimal ATP regeneration module, the utility of the proton ones (1). This incentive holds also in the case of cell membranes, pump can be extended to other applications in the context of where the scaffold phospholipids can be replaced with other synthetic cells such as transport, signaling, and control of enzy- amphiphilic molecules like synthetic polymers or blended with matic reactions. -

Curriculum Vitae KRISTI L
Curriculum Vitae KRISTI L. KIICK, PH.D. Professor www.udel.edu/mseg Department of Materials Science and Engineering [email protected] Biomedical Engineering 302 831 0201 University of Delaware, Newark, DE 19716 EDUCATION Doctor of Philosophy Polymer Science and Engineering 2001 University of Massachusetts Amherst, Amherst, MA NDSEG Predoctoral Fellow Master of Science Polymer Science and Engineering 1998 University of Massachusetts Amherst , Amherst, MA Master of Science Chemistry 1991 University of Georgia, Athens, GA NSF Predoctoral Fellow Bachelor of Science Chemistry, Summa cum laude 1989 University of Delaware, Newark, DE Eugene duPont Memorial Scholar RESEARCH INTERESTS Biologically derived methods for the synthesis and assembly of advanced macromolecular materials The development of protein engineering methods for the synthesis of artificial protein polymers functionalized with both natural and non-natural amino acids, and the chemical modification of protein polymers to produce well-defined protein-based architectures. The use of modified proteins in both biological and materials applications such as toxin neutralization, manipulation of cell signaling, and construction of light-emitting devices. The development of new elastomeric materials with well-defined conformational and assembly behavior. The assembly of hydrogel networks via protein-polysaccharide interactions to produce materials that mimic the biological activity of the extracellular matrix and that have controlled mechanical, erosion, and drug delivery properties. The -

Construction and Characterization of Hybrid
CONSTRUCTION AND CHARACTERIZATION OF HYBRID NANOPARTICLES VIA BLOCK COPOLYMER BLENDS AND KINETIC CONTROL OF SOLUTION ASSEMBLY by Yingchao Chen A dissertation submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Material Science and Engineering 2015 Spring © 2015 Yingchao Chen All Rights Reserved ProQuest Number: 3730204 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. ProQuest 3730204 Published by ProQuest LLC (2015). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, MI 48106 - 1346 CONSTRUCTION AND CHARACTERIZATION OF HYBRID NANOPARTICLES VIA BLOCK COPOLYMER BLENDS AND KINETIC CONTROL OF SOLUTION ASSEMBLY by Yingchao Chen Approved: __________________________________________________________ Darrin J. Pochan, Ph.D. Chair of the Department of Material Science and Engineering Approved: __________________________________________________________ Babatunde A. Ogunnaike, Ph.D. Dean of the College of Engineering Approved: __________________________________________________________ James G. Richards, Ph.D. Vice Provost for Graduate and Professional Education I certify that I have read this dissertation and that in my opinion it meets the academic and professional standard required by the University as a dissertation for the degree of Doctor of Philosophy. -

Smart, Biocompatible Polymersomes for Targeted Delivery of Therapeutic Agents to Cancerous Tumors
SMART, BIOCOMPATIBLE POLYMERSOMES FOR TARGETED DELIVERY OF THERAPEUTIC AGENTS TO CANCEROUS TUMORS A Dissertation Submitted to the Graduate Faculty of the North Dakota State University of Agriculture and Applied Science By Tayebeh Anajafi Marzijarani In Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Department: Pharmaceutical Sciences May 2017 Fargo, North Dakota North Dakota State University Graduate School Title SMART, BIOCOMPATIBLE POLYMERSOMES FOR TARGETED DELIVERY OF THERAPEUTIC AGENTS TO CANCEROUS TUMORS By Tayebeh Anajafi Marzijarani The Supervisory Committee certifies that this disquisition complies with North Dakota State University’s regulations and meets the accepted standards for the degree of DOCTOR OF PHILOSOPHY SUPERVISORY COMMITTEE: Dr. Sanku Mallik Chair Dr. Jagdish Singh Dr. Chengwen Sun Dr. Dean Webster Approved: 05/08/2017 Dr. Jagdish Singh Date Department Chair ABSTRACT Chemotherapeutics are the major treatment options for cancer. Although we cannot underestimate the importance of the chemotherapeutic drugs, their systemic toxicity is an important limiting factor for their use. Therefore, altering the biodistribution of the therapeutics can be an important step in treating the cancer patients. Thus, there is a growing interest in developing smart, targeted, stimuli responsive, drug delivery vehicles employing nanotechnalogy. The vehicles are often engineered to deliver the theranostics to the tumor microenvironment (extracellular matrix) or intracellular environment (e.g. cytosol, nucleus, mitochondria). The physiochemical changes in the cancerous tissues (e.g. leaky vasculature, proteins overexpression on the cell surface, overexpression of proteolytic enzymes, increased reducing agents concentration, decreased pH, and hypoxia) offer tremendous opportunities for selectively targeting the malignancies. Polymersomes are robust polymeric vesicles which have shown promising drug delivery capabilities. -

Polymersomes for Biomedical Applications: Surface Functionalization of Silicone-Based Polymer Vesicles
Polymersomes for biomedical applications: surface functionalization of silicone-based polymer vesicles Inauguraldissertation zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Stefan Egli aus Wald (ZH) Basel 2011 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel auf Antrag von Prof. Dr. Wolfgang P. Meier und Prof. Dr. Thomas Pfohl Basel, den 24. Mai 2011 Prof. Dr. Martin Spiess Dekan i „Natürlicher Verstand kann fast jeden Grad von Bildung ersetzen, aber keine Bildung den natürlichen Verstand.“ Arthur Schopenhauer (1788 – 1860) ii Acknowledgments First of all I want to thank Prof. Wolfgang Meier for giving me the opportunity to perform my PhD thesis in his research group. I admire his patience and the trust he gave me to work on my project as well as his cordiality. Also I thank Prof. Thomas Pfohl for his interest in my research work and for examining my doctoral thesis. Prof. Stefan Willitsch is kindly acknowledged for chairing the exam. Furthermore I thank our research group leaders, all our present and former Post-Doc group members. In this respect, my special thanks go to: PD Dr. Cornelia Palivan for her competent inputs, fruitful collaboration and for leading the group when times were difficult, Dr. Nico Bruns for fruitful collaboration and especially for being the greatest Queen fan ever! I also want to thank Dr. Ozana Onaca for her motivating attitude and the unfailing believe in and fight for a clean biolab, Dr. Katarzyna Kita for providing me precious advice with scientific issues. Great thanks to Dr. Per Rigler for introducing me in FCS and in the rules of publishing scientific articles. -

Polymersome Popping by Light-Induced Osmotic Shock Under
Polymersome Popping by Light-Induced Osmotic Shock under Temporal, Spatial, and Spectral Control Ariane Peyret, Emmanuel Ibarboure, Arnaud Tron, Louis Beauté, Ruben Rust, Olivier Sandre, Nathan Mcclenaghan, Sébastien Lecommandoux To cite this version: Ariane Peyret, Emmanuel Ibarboure, Arnaud Tron, Louis Beauté, Ruben Rust, et al.. Poly- mersome Popping by Light-Induced Osmotic Shock under Temporal, Spatial, and Spectral Con- trol. Angewandte Chemie International Edition, Wiley-VCH Verlag, 2017, 56 (6), pp.1566-1570. 10.1002/ange.201609231. hal-01418924 HAL Id: hal-01418924 https://hal.archives-ouvertes.fr/hal-01418924 Submitted on 9 Nov 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. COMMUNICATION Author manuscript of Angew. Chem. Int. Ed. 2017, 56 1566-1570 Polymersome popping by light-induced osmotic shock under temporal, spatial and spectral control Ariane Peyret[a], Emmanuel Ibarboure[a], Arnaud Tron[b], Louis Beauté[a], Ruben Rust[b], Olivier Sandre[a], Nathan D. McClenaghan*[b], Sebastien Lecommandoux*[a] Abstract: A high precision approach allowing light-triggered, membranes, either as a result of intrinsic membrane programmed cell-sized vesicle rupture is described, with particular permeability[23] or by the incorporation of channels or pores into emphasis on self-assembled polymersome capsules. -
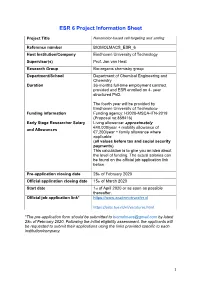
ESR 6 Project Information Sheet
ESR 6 Project Information Sheet Project Title Nanomotor-based cell targeting and sorting Reference number BIOMOLMACS_ESR_6 Host Institution/Company Eindhoven University of Technology Supervisor(s) Prof. Jan van Hest Research Group Bio-organic chemistry group Department/School Department of Chemical Engineering and Chemistry Duration 36-months full-time employment contract provided and ESR enrolled on 4- year structured PhD. The fourth year will be provided by Eindhoven University of Technology Funding information Funding agency: H2020-MSCA-ITN-2019 (Proposal no:859416) Early Stage Researcher Salary Living allowance: approximately €40,000/year + mobility allowance of and Allowances €7,200/year + family allowance where applicable (all values before tax and social security payments) This calculation is to give you an idea about the level of funding. The actual salaries can be found on the official job application link below. Pre-application closing date 28th of February 2020 Official application closing date 15th of March 2020 Start date 1st of April 2020 or as soon as possible thereafter. Official job application link* https://www.academictransfer.nl https://jobs.tue.nl/nl/vacatures.html *The pre-application form should be submitted to [email protected] by latest 28th of February 2020. Following the initial eligibility assessment, the applicants will be requested to submit their applications using the links provided specific to each institution/company. 1 Post Summary Brief description of the project: The main focus of this project will be to develop nanomotors based on polymer vesicles that can recognize, bind and isolate specific cells. Bowl-shaped vesicles, also known as stomatocytes, will be created out of biodegradable block copolymers prepared from poly(ethylene glycol)-poly(D,L- lactic acid) and loaded with enzymes, specifically glucose oxidase and catalase. -

Soft Matter PAPER
Soft Matter Dynamic Article LinksC< Cite this: Soft Matter, 2012, 8, 3810 www.rsc.org/softmatter PAPER Electrodeformation method for measuring the capacitance of bilayer membranes Paul F. Salipante,ab Roland L. Knorr,b Rumiana Dimovab and Petia M. Vlahovska*a Received 4th November 2011, Accepted 23rd January 2012 DOI: 10.1039/c2sm07105c We report a novel non-invasive method to measure the capacitance of biomimetic bilayer membranes. The approach utilizes the frequency-dependent deformation of giant unilamellar vesicles in AC uniform electric field. The method is applied to membranes made of lipids and polymers. Compared to lipid membranes, polymer bilayers have an order of magnitude lower capacitance, which correlates with their larger thickness. The capacitance of both lipid and polymer bilayers is found to be voltage- independent in the examined range between 2 and 6 kV mÀ1, indicating membranes do not experience significant thinning due to electrostriction. 1 Introduction real cells include classic electrophysiological techniques such as patch-clamp,10 electromechanical methods based on individual Electric properties of biomembranes play a pivotal role in cell dielectrophoresis, electrorotation and electro-orientation,11 cellular functions,1 e.g., synaptic efficacy and electric signal 2–4 5 and dielectric spectroscopy based on the frequency-dependent propagation in neurons, as well as biomedical applications, permittivity and conductivity of suspensions of cells12–14 or e.g., gene transfection, where the controlled application of elec- liposomes.15 The patch-clamp method is very effective, but can tric pulses induces transient pores in the cell membrane thereby be invasive and laborious. Dielectric spectroscopy is non-inva- enabling the delivery of foreign DNA into the cell. -

PDF Hosted at the Radboud Repository of the Radboud University Nijmegen
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/161485 Please be advised that this information was generated on 2017-12-06 and may be subject to change. Engineering compartmentalised life-like molecular systems Marlies Nijemeisland Engineering compartmentalised life-like molecular systems Proefschrift ter verkrijging van de graad van doctor aan de Radboud Universiteit Nijmegen op gezag van de rector magnificus prof. dr. J.H.J.M. van Krieken, volgens besluit van het college van decanen in het openbaar te verdedigen op donderdag 12 januari 2017 om 12.30 uur precies door Marlies Nijemeisland geboren op 22 september 1984 te Zelhem Promotoren: Prof. dr. ir. Jan C.M. van Hest Prof. dr. Wilhelm T.S. Huck Manuscriptcommissie: Prof. dr. Roeland J.M. Nolte (voorzitter) Prof. dr. Jan H. van Esch (Technische Universiteit Delft) Dr. ir. Tom F.A. de Greef (Technische Universiteit Eindhoven) ISBN: 978-94-92380-06-7 © 2016 Marlies Nijemeisland This work was financially supported by the Radboud University (Bionic Cell project), the Ministry of Education, Culture and Science (Gravitation program 024.001.035) and funding from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-20012)/ERC- StG 307679 “StomaMotors”. Cover design: Ria Nijemeisland-Heusinkveld Press: Gildeprint Table of Contents Chapter 1 General introduction and thesis outline ......................................... 1 1.1 Introduction ........................................................................................... 2 1.2 Compartments ........................................................................................ 2 1.2.1 Membrane vesicles ....................................................................... 3 1.2.2 Multi-compartment systems ........................................................ -

Artificial Cells: Synthetic Compartments with Life-Like Functionality and Adaptivity
This is an open access article published under a Creative Commons Non-Commercial No Derivative Works (CC-BY-NC-ND) Attribution License, which permits copying and redistribution of the article, and creation of adaptations, all for non-commercial purposes. Article pubs.acs.org/accounts Artificial Cells: Synthetic Compartments with Life-like Functionality and Adaptivity Bastiaan C. Buddingh’ and Jan C. M. van Hest* Eindhoven University of Technology, P.O. Box 513 (STO 3.31), 5600 MB Eindhoven, The Netherlands CONSPECTUS: Cells are highly advanced microreactors that form the basis of all life. Their fascinating complexity has inspired scientists to create analogs from synthetic and natural components using a bottom-up approach. The ultimate goal here is to assemble a fully man-made cell that displays functionality and adaptivity as advanced as that found in nature, which will not only provide insight into the fundamental processes in natural cells but also pave the way for new applications of such artificial cells. In this Account, we highlight our recent work and that of others on the construction of artificial cells. First, we will introduce the key features that characterize a living system; next, we will discuss how these have been imitated in artificial cells. First, compartmentalization is crucial to separate the inner chemical milieu from the external environment. Current state-of-the-art artificial cells comprise subcompartments to mimic the hierarchical architecture of eukaryotic cells and tissue. Furthermore, synthetic gene circuits have been used to encode genetic information that creates complex behavior like pulses or feedback. Additionally, artificial cells have to reproduce to maintain a population. -

Meet the Editorial Board DOI: 10.1039/C001033m
PROFILE www.rsc.org/polymers | Polymer Chemistry Meet the Editorial Board DOI: 10.1039/c001033m Professor David Haddleton, for Polymer Research, obtaining her PhD a Member of the Chinese Academy of Editor-in-Chief in 1998 for work in the area of fullerene Sciences. adducts and polymers. A postdoctoral Professor Academician Yang was fellowship with CPIMA (NSF-Center for a faculty member of Fudan University Polymer Interfaces and Macromolecular before he was appointed Vice President Assemblies) brought her to the IBM for Research in 1999. In 2006, he was Almaden Research Center, California appointed Director General of the USA, to work under the direction of Prof. Department of Degree and Postgraduate Craig J. Hawker focusing on the devel- Education, State Council of the People’s opment of new living free polymerization Republic of China. Professor Academi- techniques and approaches to nanoscopic cian Yang serves as senior advisor to the materials. In 2001 she joined XenoPort, Shanghai Municipal Government. He Inc. as a Staff Scientist investigating was inaugurated as President of Fudan enabling technologies for the increased University in January 2009. bioavailability of macromolecular thera- The academic interests of Professor peutics. After this extensive industrial Academician Yang are in condensed experience she started at Vanderbilt matter physics and polymer science. He David Haddleton is Professor of Chem- University as Assistant Professor in the owns many domestic and international istry at the University of Warwick, UK. Department of Chemistry in 2004 with patents and serves on the boards of Professor Haddleton’s research centres a secondary appointment in the Depart- several academic journals including on controlled polymerisation to give ment of Pharmacology, Vanderbilt Chinese Journal of Chemistry and Science macromolecules of designed, desired and Medical School.