The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enhanced Representation of Natural Product Metabolism in Uniprotkb
H OH metabolites OH Article Diverse Taxonomies for Diverse Chemistries: Enhanced Representation of Natural Product Metabolism in UniProtKB Marc Feuermann 1,* , Emmanuel Boutet 1,* , Anne Morgat 1 , Kristian B. Axelsen 1, Parit Bansal 1, Jerven Bolleman 1 , Edouard de Castro 1, Elisabeth Coudert 1, Elisabeth Gasteiger 1,Sébastien Géhant 1, Damien Lieberherr 1, Thierry Lombardot 1,†, Teresa B. Neto 1, Ivo Pedruzzi 1, Sylvain Poux 1, Monica Pozzato 1, Nicole Redaschi 1 , Alan Bridge 1 and on behalf of the UniProt Consortium 1,2,3,4,‡ 1 Swiss-Prot Group, SIB Swiss Institute of Bioinformatics, CMU, 1 Michel-Servet, CH-1211 Geneva 4, Switzerland; [email protected] (A.M.); [email protected] (K.B.A.); [email protected] (P.B.); [email protected] (J.B.); [email protected] (E.d.C.); [email protected] (E.C.); [email protected] (E.G.); [email protected] (S.G.); [email protected] (D.L.); [email protected] (T.L.); [email protected] (T.B.N.); [email protected] (I.P.); [email protected] (S.P.); [email protected] (M.P.); [email protected] (N.R.); [email protected] (A.B.); [email protected] (U.C.) 2 European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SD, UK 3 Protein Information Resource, University of Delaware, 15 Innovation Way, Suite 205, Newark, DE 19711, USA 4 Protein Information Resource, Georgetown University Medical Center, 3300 Whitehaven Street NorthWest, Suite 1200, Washington, DC 20007, USA * Correspondence: [email protected] (M.F.); [email protected] (E.B.); Tel.: +41-22-379-58-75 (M.F.); +41-22-379-49-10 (E.B.) † Current address: Centre Informatique, Division Calcul et Soutien à la Recherche, University of Lausanne, CH-1015 Lausanne, Switzerland. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

Research Resources for Nuclear Receptor Signaling Pathways Neil J
Molecular Pharmacology Fast Forward. Published on May 23, 2016 as DOI: 10.1124/mol.116.103713 This article has not been copyedited and formatted. The final version may differ from this version. MOL #103713 Research resources for nuclear receptor signaling pathways Neil J. McKenna Department of Molecular and Cellular Biology and Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource, Downloaded from Baylor College of Medicine, Houston, TX, 77030, USA molpharm.aspetjournals.org at ASPET Journals on September 27, 2021 1 Molecular Pharmacology Fast Forward. Published on May 23, 2016 as DOI: 10.1124/mol.116.103713 This article has not been copyedited and formatted. The final version may differ from this version. MOL #103713 Running title: Research resources for NR signaling pathways Corresponding author: Neil J McKenna Room M620 Baylor College of Medicine One Baylor Plaza Downloaded from Houston, TX, 77030, USA t: 713-798-7490 molpharm.aspetjournals.org f: 713-798-6822 e: [email protected] Number of text pages: 21 at ASPET Journals on September 27, 2021 Number of tables: 1 Number of figures: 1 Number of references: 56 Number of words in Abstract: 124 Review: 3613 List of non-standard abbreviations: 17βE2, 17β-estradiol; AB, Allen Brain Atlas; BG, BIOGRID; BGS, BioGPS; CoR, coregulator; CTD, Comparative Toxicogenomics Database; DAV, DAVID; DB, DrugBank; EDC, endocrine disrupting chemical; EG, Entrez Gene; EM, Edinburgh Mouse; ENC, ENCODE; ENR, ENRICHR; ENS, Ensembl; EX, Expression Atlas; GC, GeneCards; GSEA, GeneSet Enrichment Analysis; GtoP, IUPHAR Guide To Pharmacology; 2 Molecular Pharmacology Fast Forward. Published on May 23, 2016 as DOI: 10.1124/mol.116.103713 This article has not been copyedited and formatted. -

Molecular Structure of Promoter-Bound Yeast TFIID
ARTICLE DOI: 10.1038/s41467-018-07096-y OPEN Molecular structure of promoter-bound yeast TFIID Olga Kolesnikova 1,2,3,4, Adam Ben-Shem1,2,3,4, Jie Luo5, Jeff Ranish 5, Patrick Schultz 1,2,3,4 & Gabor Papai 1,2,3,4 Transcription preinitiation complex assembly on the promoters of protein encoding genes is nucleated in vivo by TFIID composed of the TATA-box Binding Protein (TBP) and 13 TBP- associate factors (Tafs) providing regulatory and chromatin binding functions. Here we present the cryo-electron microscopy structure of promoter-bound yeast TFIID at a resolu- 1234567890():,; tion better than 5 Å, except for a flexible domain. We position the crystal structures of several subunits and, in combination with cross-linking studies, describe the quaternary organization of TFIID. The compact tri lobed architecture is stabilized by a topologically closed Taf5-Taf6 tetramer. We confirm the unique subunit stoichiometry prevailing in TFIID and uncover a hexameric arrangement of Tafs containing a histone fold domain in the Twin lobe. 1 Department of Integrated Structural Biology, Equipe labellisée Ligue Contre le Cancer, Institut de Génétique et de Biologie Moléculaire et Cellulaire, Illkirch 67404, France. 2 Centre National de la Recherche Scientifique, UMR7104, 67404 Illkirch, France. 3 Institut National de la Santé et de la Recherche Médicale, U1258, 67404 Illkirch, France. 4 Université de Strasbourg, Illkirch 67404, France. 5 Institute for Systems Biology, Seattle, WA 98109, USA. These authors contributed equally: Olga Kolesnikova, Adam Ben-Shem. Correspondence and requests for materials should be addressed to P.S. (email: [email protected]) or to G.P. -

Uniprot: the Universal Protein Knowledgebase in 2021 the Uniprot Consortium1,2,3,4,*
D480–D489 Nucleic Acids Research, 2021, Vol. 49, Database issue Published online 25 November 2020 doi: 10.1093/nar/gkaa1100 UniProt: the universal protein knowledgebase in 2021 The UniProt Consortium1,2,3,4,* 1European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Hinxton CB10 1SD, UK, 2Protein Information Resource, Georgetown University Medical Center, 3300 Whitehaven Street NW, Suite 1200, Washington, DC 20007, USA, 3Protein Information Resource, University of Delaware, Ammon-Pinizzotto Biopharmaceutical Innovation Building, Suite 147, 590 Avenue 1743, Newark, DE 19713, USA and 4SIB Swiss Institute of Bioinformatics, Centre Medical Universitaire, 1 rue Michel Servet, CH-1211 Geneva 4, Switzerland Received September 15, 2020; Revised October 21, 2020; Editorial Decision October 22, 2020; Accepted November 02, 2020 ABSTRACT tomated systems. The UniRef databases cluster sequence sets at various levels of sequence identity and the UniProt The aim of the UniProt Knowledgebase is to provide Archive (UniParc) delivers a complete set of known se- users with a comprehensive, high-quality and freely quences, including historical obsolete sequences. UniProt accessible set of protein sequences annotated with additionally integrates, interprets, and standardizes data functional information. In this article, we describe from multiple selected resources to add biological knowl- significant updates that we have made over the last edge and associated metadata to protein records and acts two years to the resource. The number of sequences as a central hub from which users can link out to 180 in UniProtKB has risen to approximately 190 million, other resources. In recognition of the quality of our data, despite continued work to reduce sequence redun- and the service we provide, UniProt was recognised as dancy at the proteome level. -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

Molecular Mechanisms of Ribosomal Protein Gene Coregulation
Downloaded from genesdev.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press Molecular mechanisms of ribosomal protein gene coregulation Rohit Reja, Vinesh Vinayachandran, Sujana Ghosh, and B. Franklin Pugh Center for Eukaryotic Gene Regulation, Pennsylvania State University, University Park, Pennsylvania 16802, USA The 137 ribosomal protein genes (RPGs) of Saccharomyces provide a model for gene coregulation. We examined the positional and functional organization of their regulators (Rap1 [repressor activator protein 1], Fhl1, Ifh1, Sfp1, and Hmo1), the transcription machinery (TFIIB, TFIID, and RNA polymerase II), and chromatin at near-base-pair res- olution using ChIP-exo, as RPGs are coordinately reprogrammed. Where Hmo1 is enriched, Fhl1, Ifh1, Sfp1, and Hmo1 cross-linked broadly to promoter DNA in an RPG-specific manner and demarcated by general minor groove widening. Importantly, Hmo1 extended 20–50 base pairs (bp) downstream from Fhl1. Upon RPG repression, Fhl1 remained in place. Hmo1 dissociated, which was coupled to an upstream shift of the +1 nucleosome, as reflected by the Hmo1 extension and core promoter region. Fhl1 and Hmo1 may create two regulatable and positionally distinct barriers, against which chromatin remodelers position the +1 nucleosome into either an activating or a repressive state. Consistent with in vitro studies, we found that specific TFIID subunits, in addition to cross-linking at the core promoter, made precise cross-links at Rap1 sites, which we interpret to reflect native Rap1–TFIID interactions. Our findings suggest how sequence-specific DNA binding regulates nucleosome positioning and transcription complex assembly >300 bp away and how coregulation coevolved with coding sequences. -

The Chromosome-Centric Human Proteome Project for Cataloging Proteins Encoded in the Genome
CORRESPONDENCE The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome To the Editor: utility for biological and disease studies. Table 1 Features of salient genes on The Chromosome-Centric Human With development of new tools for in- chromosomes 13 and 17 Proteome Project (C-HPP) aims to define depth characterization of the transcriptome Genea AST nsSNPs the full set of proteins encoded in each and proteome, the HPP is well positioned Chromosome 13 chromosome through development of a to have a strategic role in addressing the BRCA2 3 54 standardized approach for analyzing the complexity of human phenotypes. With this RB1 2 3 massive proteomic data sets currently being in mind, the HUPO has organized national IRS2 1 3 generated from dedicated efforts of national chromosome teams that will collaborate and international teams. The initial goal with well-established laboratories building Chromosome 17 of the C-HPP is to identify at least one complementary proteotypic peptides, BRCA1 24 24 representative protein encoded by each of antibodies and informatics resources. ERBB2 6 13 the approximately 20,300 human genes1,2. An important C-HPP goal is to encourage TP53 14 5 aEnsembl protein and AST information can be found at The proteins will be characterized for tissue capture and open sharing of proteomic http://www.ensembl.org/Homo_sapiens/. localization and major isoforms, including data sets from diverse samples to enhance AST, alternative splicing transcript; nsSNP, nonsyno- mous single-nucleotide polyphorphism assembled from post-translational modifications (PTMs), a gene- and chromosome-centric display data from the 1000 Genomes Projects. -

TAF10 Complex Provides Evidence for Nuclear Holo&Ndash;TFIID Assembly from Preform
ARTICLE Received 13 Aug 2014 | Accepted 2 Dec 2014 | Published 14 Jan 2015 DOI: 10.1038/ncomms7011 OPEN Cytoplasmic TAF2–TAF8–TAF10 complex provides evidence for nuclear holo–TFIID assembly from preformed submodules Simon Trowitzsch1,2, Cristina Viola1,2, Elisabeth Scheer3, Sascha Conic3, Virginie Chavant4, Marjorie Fournier3, Gabor Papai5, Ima-Obong Ebong6, Christiane Schaffitzel1,2, Juan Zou7, Matthias Haffke1,2, Juri Rappsilber7,8, Carol V. Robinson6, Patrick Schultz5, Laszlo Tora3 & Imre Berger1,2,9 General transcription factor TFIID is a cornerstone of RNA polymerase II transcription initiation in eukaryotic cells. How human TFIID—a megadalton-sized multiprotein complex composed of the TATA-binding protein (TBP) and 13 TBP-associated factors (TAFs)— assembles into a functional transcription factor is poorly understood. Here we describe a heterotrimeric TFIID subcomplex consisting of the TAF2, TAF8 and TAF10 proteins, which assembles in the cytoplasm. Using native mass spectrometry, we define the interactions between the TAFs and uncover a central role for TAF8 in nucleating the complex. X-ray crystallography reveals a non-canonical arrangement of the TAF8–TAF10 histone fold domains. TAF2 binds to multiple motifs within the TAF8 C-terminal region, and these interactions dictate TAF2 incorporation into a core–TFIID complex that exists in the nucleus. Our results provide evidence for a stepwise assembly pathway of nuclear holo–TFIID, regulated by nuclear import of preformed cytoplasmic submodules. 1 European Molecular Biology Laboratory, Grenoble Outstation, 6 rue Jules Horowitz, 38042 Grenoble, France. 2 Unit for Virus Host-Cell Interactions, University Grenoble Alpes-EMBL-CNRS, 6 rue Jules Horowitz, 38042 Grenoble, France. 3 Cellular Signaling and Nuclear Dynamics Program, Institut de Ge´ne´tique et de Biologie Mole´culaire et Cellulaire, UMR 7104, INSERM U964, 1 rue Laurent Fries, 67404 Illkirch, France. -

MAF1 Represses CDKN1A Through a Pol III-Dependent Mechanism Yu-Ling Lee, Yuan-Ching Li, Chia-Hsin Su, Chun-Hui Chiao, I-Hsuan Lin, Ming-Ta Hsu*
RESEARCH ARTICLE elifesciences.org MAF1 represses CDKN1A through a Pol III-dependent mechanism Yu-Ling Lee, Yuan-Ching Li, Chia-Hsin Su, Chun-Hui Chiao, I-Hsuan Lin, Ming-Ta Hsu* Institute of Biochemistry and Molecular Biology, School of Life Science, National Yang-Ming University, Taipei, Taiwan Abstract MAF1 represses Pol III-mediated transcription by interfering with TFIIIB and Pol III. Herein, we found that MAF1 knockdown induced CDKN1A transcription and chromatin looping concurrently with Pol III recruitment. Simultaneous knockdown of MAF1 with Pol III or BRF1 (subunit of TFIIIB) diminished the activation and looping effect, which indicates that recruiting Pol III was required for activation of Pol II-mediated transcription and chromatin looping. Chromatin- immunoprecipitation analysis after MAF1 knockdown indicated enhanced binding of Pol III and BRF1, as well as of CFP1, p300, and PCAF, which are factors that mediate active histone marks, along with the binding of TATA binding protein (TBP) and POLR2E to the CDKN1A promoter. Simultaneous knockdown with Pol III abolished these regulatory events. Similar results were obtained for GDF15. Our results reveal a novel mechanism by which MAF1 and Pol III regulate the activity of a protein- coding gene transcribed by Pol II. DOI: 10.7554/eLife.06283.001 Introduction Transcription by RNA polymerase III (Pol III) is regulated by MAF1, which is a highly conserved protein in eukaryotes (Pluta et al., 2001; Reina et al., 2006). MAF1 represses Pol III transcription through *For correspondence: mth@ym. association with BRF1, a subunit of initiation factor TFIIIB, which prevents attachment of TFIIIB onto edu.tw DNA. -
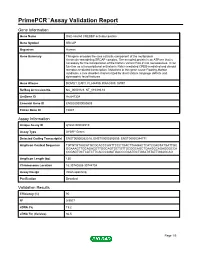
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name Snf2-related CREBBP activator protein Gene Symbol SRCAP Organism Human Gene Summary This gene encodes the core catalytic component of the multiprotein chromatin-remodeling SRCAP complex. The encoded protein is an ATPase that is necessary for the incorporation of the histone variant H2A.Z into nucleosomes. It can function as a transcriptional activator in Notch-mediated CREB-mediated and steroid receptor-mediated transcription. Mutations in this gene cause Floating-Harbor syndrome a rare disorder characterized by short stature language deficits and dysmorphic facial features. Gene Aliases DOMO1, EAF1, FLJ44499, KIAA0309, SWR1 RefSeq Accession No. NC_000016.9, NT_010393.16 UniGene ID Hs.647334 Ensembl Gene ID ENSG00000080603 Entrez Gene ID 10847 Assay Information Unique Assay ID qHsaCID0006510 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000262518, ENST00000395059, ENST00000344771 Amplicon Context Sequence TGTGTGTAACATGCGCACCCAGTTCCCTGACTTAAGACTCATCCAGTATGATTGC GGAAAGTTGCAGACGTTGGCAGTGCTGTTGCGGCAGCTCAAGGCAGAGGGCCA CCGAGTGCTCATCTTCACCCAGATGACCCGAATGCTGGATGTATTGGAGCAG Amplicon Length (bp) 130 Chromosome Location 16:30740838-30744704 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 90 R2 0.9977 cDNA Cq 19.2 cDNA Tm (Celsius) 86.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report SRCAP, Human -

How Many Human Proteoforms Are There?
PERSPECTIVE PUBLISHED ONLINE: 14 FEBRUARY 2018 | DOI: 10.1038/NCHEMBIO.2576 How many human proteoforms are there? Ruedi Aebersold1, Jeffrey N Agar2, I Jonathan Amster3 , Mark S Baker4 , Carolyn R Bertozzi5, Emily S Boja6, Catherine E Costello7, Benjamin F Cravatt8 , Catherine Fenselau9, Benjamin A Garcia10, Ying Ge11,12, Jeremy Gunawardena13, Ronald C Hendrickson14, Paul J Hergenrother15, Christian G Huber16 , Alexander R Ivanov2, Ole N Jensen17, Michael C Jewett18, Neil L Kelleher19* , Laura L Kiessling20 , Nevan J Krogan21, Martin R Larsen17, Joseph A Loo22 , Rachel R Ogorzalek Loo22, Emma Lundberg23,24, Michael J MacCoss25, Parag Mallick5, Vamsi K Mootha13, Milan Mrksich18, Tom W Muir26, Steven M Patrie19, James J Pesavento27 , Sharon J Pitteri5 , Henry Rodriguez6, Alan Saghatelian28, Wendy Sandoval29, Hartmut Schlüter30 , Salvatore Sechi31, Sarah A Slavoff32, Lloyd M Smith12,33, Michael P Snyder24, Paul M Thomas19 , Mathias Uhlén34, Jennifer E Van Eyk35, Marc Vidal36, David R Walt37, Forest M White38, Evan R Williams39, Therese Wohlschlager16, Vicki H Wysocki40, Nathan A Yates41, Nicolas L Young42 & Bing Zhang42 Despite decades of accumulated knowledge about proteins and their post-translational modifications (PTMs), numerous ques- tions remain regarding their molecular composition and biological function. One of the most fundamental queries is the extent to which the combinations of DNA-, RNA- and PTM-level variations explode the complexity of the human proteome. Here, we outline what we know from current databases and measurement strategies including mass spectrometry–based proteomics. In doing so, we examine prevailing notions about the number of modifications displayed on human proteins and how they combine to generate the protein diversity underlying health and disease.