Unix Activities – Week 1 Assignment 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Application for a Certificate of Eligibility to Employ Child Performers
Division of Labor Standards Permit and Certificate Unit Harriman State Office Campus Building 12, Room 185B Albany, NY 12240 www.labor.ny.gov Application for a Certificate of Eligibility to Employ Child Performers A. Submission Instructions A Certificate of Eligibility to Employ Child Performers must be obtained prior to employing any child performer. Certificates are renew able every three (3) years. To obtain or renew a certificate: • Complete Parts B, C, D and E of this application. • Attach proof of New York State Workers’ Compensation and Disability Insurance. o If you currently have employees in New York, you must provide proof of coverage for those New York State w orkers by attaching copies of Form C-105.2 and DB-120.1, obtainable from your insurance carrier. o If you are currently exempt from this requirement, complete Form CE-200 attesting that you are not required to obtain New York State Workers’ Compensation and Disability Insurance Coverage. Information on and copies of this form are available from any district office of the Workers’ Compensation Board or from their w ebsite at w ww.wcb.ny.gov, Click on “WC/DB Exemptions,” then click on “Request for WC/DB Exemptions.” • Attach a check for the correct amount from Section D, made payable to the Commissioner of Labor. • Sign and mail this completed application and all required documents to the address listed above. If you have any questions, call (518) 457-1942, email [email protected] or visit the Department’s w ebsite at w ww.labor.ny.gov B. Type of Request (check one) New Renew al Current certificate number _______________________________________________ Are you seeking this certificate to employ child models? Yes No C. -

Program #6: Word Count
CSc 227 — Program Design and Development Spring 2014 (McCann) http://www.cs.arizona.edu/classes/cs227/spring14/ Program #6: Word Count Due Date: March 11 th, 2014, at 9:00 p.m. MST Overview: The UNIX operating system (and its variants, of which Linux is one) includes quite a few useful utility programs. One of those is wc, which is short for Word Count. The purpose of wc is to give users an easy way to determine the size of a text file in terms of the number of lines, words, and bytes it contains. (It can do a bit more, but that’s all of the functionality that we are concerned with for this assignment.) Counting lines is done by looking for “end of line” characters (\n (ASCII 10) for UNIX text files, or the pair \r\n (ASCII 13 and 10) for Windows/DOS text files). Counting words is also straight–forward: Any sequence of characters not interrupted by “whitespace” (spaces, tabs, end–of–line characters) is a word. Of course, whitespace characters are characters, and need to be counted as such. A problem with wc is that it generates a very minimal output format. Here’s an example of what wc produces on a Linux system when asked to count the content of a pair of files; we can do better! $ wc prog6a.dat prog6b.dat 2 6 38 prog6a.dat 32 321 1883 prog6b.dat 34 327 1921 total Assignment: Write a Java program (completely documented according to the class documentation guidelines, of course) that counts lines, words, and bytes (characters) of text files. -
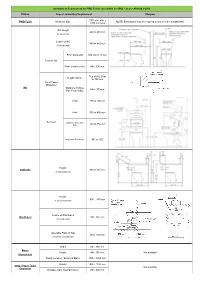
Summary of Requirement for PWD Toilets (Not Within an SOU) – As Per AS1428.1-2009
Summary of Requirement for PWD Toilets (not within an SOU) – as per AS1428.1-2009 Fixture Aspect and/or Key Requirement Diagram 1900 mm wide x PWD Toilet Minimum Size NOTE: Extra space may be required to allow for the a washbasin. 2300 mm long WC Height 460 to 480 mm (to top of seat) Centre of WC 450 to 460 mm (from side wall) From back wall 800 mm ± 10 mm Front of WC From cistern or like Min. 600 mm Top of WC Seat Height of Zone to 700 mm Toilet Paper Dispenser WC Distance of Zone Max. 300mm from Front of QC Height 150 to 200 mm Width 350 to 400 mm Backrest Distance above WC Seat 120 to 150 mm Angle from Seat Hinge 90° to 100° Height Grabrails 800 to 810 mm (to top of grab rail) Height 800 – 830 mm (to top of washbasin) Centre of Washbasin Washbasin Min. 425 mm (from side wall) Operable Parts of Tap Max. 300 mm (from front of washbasin) Width Min. 350 mm Mirror Height Min. 950 mm Not available (if provided) Fixing Location / Extent of Mirror 900 – 1850 mm Height 900 – 1100 mm Soap / Paper Towel Not available Dispenser Distance from Internal Corner Min. 500 mm Summary of Requirement for PWD Toilets (not within an SOU) – as per AS1428.1-2009 Fixture Aspect and/or Key Requirement Diagram Height 1200 – 1350 mm PWD Clothes Hook Not available Distance from Internal Corner Min. 500 mm Height 800 – 830 mm As vanity bench top Width Min. 120 mm Depth 300 – 400 mm Shelves Not available Height 900 – 1000 mm As separate fixture (if within any required Width 120 – 150 mm circulation space) Length 300 – 400 mm Width Min. -

Useful Commands in Linux and Other Tools for Quality Control
Useful commands in Linux and other tools for quality control Ignacio Aguilar INIA Uruguay 05-2018 Unix Basic Commands pwd show working directory ls list files in working directory ll as before but with more information mkdir d make a directory d cd d change to directory d Copy and moving commands To copy file cp /home/user/is . To copy file directory cp –r /home/folder . to move file aa into bb in folder test mv aa ./test/bb To delete rm yy delete the file yy rm –r xx delete the folder xx Redirections & pipe Redirection useful to read/write from file !! aa < bb program aa reads from file bb blupf90 < in aa > bb program aa write in file bb blupf90 < in > log Redirections & pipe “|” similar to redirection but instead to write to a file, passes content as input to other command tee copy standard input to standard output and save in a file echo copy stream to standard output Example: program blupf90 reads name of parameter file and writes output in terminal and in file log echo par.b90 | blupf90 | tee blup.log Other popular commands head file print first 10 lines list file page-by-page tail file print last 10 lines less file list file line-by-line or page-by-page wc –l file count lines grep text file find lines that contains text cat file1 fiel2 concatenate files sort sort file cut cuts specific columns join join lines of two files on specific columns paste paste lines of two file expand replace TAB with spaces uniq retain unique lines on a sorted file head / tail $ head pedigree.txt 1 0 0 2 0 0 3 0 0 4 0 0 5 0 0 6 0 0 7 0 0 8 0 0 9 0 0 10 -

Install and Run External Command Line Softwares
job monitor and control top: similar to windows task manager (space to refresh, q to exit) w: who is there ps: all running processes, PID, status, type ps -ef | grep yyin bg: move current process to background fg: move current process to foreground jobs: list running and suspended processes kill: kill processes kill pid (could find out using top or ps) 1 sort, cut, uniq, join, paste, sed, grep, awk, wc, diff, comm, cat All types of bioinformatics sequence analyses are essentially text processing. Unix Shell has the above commands that are very useful for processing texts and also allows the output from one command to be passed to another command as input using pipe (“|”). less cosmicRaw.txt | cut -f2,3,4,5,8,13 | awk '$5==22' | cut -f1 | sort -u | wc This makes the processing of files using Shell very convenient and very powerful: you do not need to write output to intermediate files or load all data into the memory. For example, combining different Unix commands for text processing is like passing an item through a manufacturing pipeline when you only care about the final product 2 Hands on example 1: cosmic mutation data - Go to UCSC genome browser website: http://genome.ucsc.edu/ - On the left, find the Downloads link - Click on Human - Click on Annotation database - Ctrl+f and then search “cosmic” - On “cosmic.txt.gz” right-click -> copy link address - Go to the terminal and wget the above link (middle click or Shift+Insert to paste what you copied) - Similarly, download the “cosmicRaw.txt.gz” file - Under your home, create a folder -

CS2043 - Unix Tools & Scripting Cornell University, Spring 20141
CS2043 - Unix Tools & Scripting Cornell University, Spring 20141 Instructor: Bruno Abrahao January 31, 2014 1 Slides evolved from previous versions by Hussam Abu-Libdeh and David Slater Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting Vim: Tip of the day! Line numbers Displays line number in Vim: :set nu Hides line number in Vim: :set nonu Goes to line number: :line number Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting Counting wc How many lines of code are in my new awesome program? How many words are in this document? Good for bragging rights Word, Character, Line, and Byte count with wc wc -l : count the number of lines wc -w : count the number of words wc -m : count the number of characters wc -c : count the number of bytes Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting Sorting sort Sorts the lines of a text file alphabetically. sort -ru file sorts the file in reverse order and deletes duplicate lines. sort -n -k 2 -t : file sorts the file numerically by using the second column, separated by a colon Example Consider a file (numbers.txt) with the numbers 1, 5, 8, 11, 62 each on a separate line, then: $ sort numbers.txt $ sort numbers.txt -n 1 1 11 5 5 8 62 11 8 62 Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting uniq uniq uniq file - Discards all but one of successive identical lines uniq -c file - Prints the number of successive identical lines next to each line Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting Character manipulation! The Translate Command tr [options] <char_list1> [char_list2] Translate or delete characters char lists are strings of characters By default, searches for characters in char list1 and replaces them with the ones that occupy the same position in char list2 Example: tr 'AEIOU' 'aeiou' - changes all capital vowels to lower case vowels Instructor: Bruno Abrahao CS2043 - Unix Tools & Scripting Pipes and redirection tr only receives input from standard input (stdin) i.e. -

Online Meeting List
PHONE NUMBERS ONLINE MEETING LIST OCTOBER 2021 24 HOUR HELPLINE 1-800-317-3222 Meeting Format Legend BEG Beginners BT Basic Text C Closed CAN Candlelight CW Children Welcome DISC Discussion IP IP Study JFT Just for Today Study LIT Literature Study M Men NC No Children O Open QA Question and Answer RA Restricted Attendance SPK Speaker STEP Step SWG Step Working Guide Study TC Temporary Closed https://freestatena.org TOP Topic TRAD Tradition VAR Format Varies VM Virtual Web Meeting SUGGESTIONS FOR EVERYONE W Women WC ♿ Wheelchair DON'T USE no matter what Ask your Higher Power to keep you clean Come early and stay late Get a home group Go to 90 meetings in 90 days Read NA literature daily Get and use a sponsor Use the PHONE KEEP COMING BACK. IT WORKS Page 11 Page 1 SUNDAY / DOMINGO Anne Arundel 7:00PM 1.25 Live and Learn, Annapolis, MD, 21401 HRS https://zoom.us/j/215757961 (O,SPK,WC ♿,VM) Baltimore 10:00AM 1 HRS Step Into The Light, Loch Raven & Taylor Ave -- meeting is in small chapel behind church, Baltimore, MD, 21286 https://zoom.us/j/836486057 Zoom ID: 836 486 057 (O,VAR,VM,LIT) 8:00PM 1 HRS Liberty Crossing, Randallstown, MD, 21133 https://us02web.zoom.us/j/6626633629?pwd=UDQvY3BBdERIQkREaytmWE1KV3l5QT09 (O,WC ♿,TC,VM) Baltimore City 10:00AM 1 HRS Literature Is the Program, Baltimore, MD, 21218 https://us04web.zoom.us/j/349786148?pwd=NjBWbHdmWlVxQ0l0SVdjVFdTWTd1UT09 Zoom ID: 349 786 148 Password: 008895 (O,BT,JFT,VAR,STEP,TRAD,VM) 1st Sunday-Step, 2nd-Tradition, 3rd-Text Study, 4th-Service & Committment, 5th-Just for Today 11:30AM -
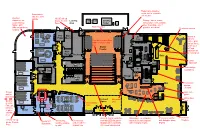
LS Lab Women's WC Men's WC Vestibule School Store Office
Retractable bleacher seats, not to encroach Greenhouse into theater and access to Outdoor outside 10’x10’ roll-up classroom for doors, French Loading Electrical Cooling Storage: tables, chairs, Lower School: Porte Area Equip Tower school store, cafe, washer, Nature path, dryer, backstage use if Main Projector different surfaces, possible, a/v racks? Check-out counter seating Fire Main AV Variety of Pump LS Lab Mech. Clos. stations: Seat finish match floor, UP Coffee, grab fabric on bottom and and go items, back combi-oven Studio hot food, Theater G R induction for Main live food prep Elect. LS Lab Backstage Backstage Cafe Catering Large Kitchen Aquarium Women's Elec. Vest. Corridor WC Grab and go Approx 200 Seats Total M Elev. food/drinks Classroom Elec. Data M Storage Office Men's Retractable bleacher Stair WC seats, not to encroach into theater Stair Front-back Classroom access School elevator Store Screen M Storefront wall for C glass distractions M Information Break-out Storefront C Classroom Desk Outdoor Space glass Shop Maker's Glass display area for Space Fab Lab, Sound- racks concrete proof walls or paver Metals Vestibule exterior area Wood & Digital Exhibition Stand-alone merchandise Check-out counter Ventless 10’x10’ roll-up Fireplace Meets MIT Tool storage, Cart storage wall with LED flat panel and built-in wall displays with lockable glass doors, French Standards lockable storage underneath for displays on metal tracks with changing rooms display Porte for staff student use with wood background 8 Fume hoods, (4 on 2nd floor, -

Product Information (PI)
Product Information Rechargeable Nickel-Metal-Hydride Button Cells 55602 506 019 6 / V 15 H LS S WC Page 1 / 2 System highlights of Ni-MH Button Cells – Robust Family – from VARTA Microbattery: Excellent high-rate discharge characteristics (3 CA / 5 CA). For short duration even higher currents can be drained No memory effect Long life – typical 500 full cycles Good overcharge capability Low self-discharge Flat discharge voltage Electrical characteristics*: Slim design Nominal Voltage: 7.2 V Wide temperature range -Storage: -40 ˚C up to +65 ˚C Typical Capacity: 15 mAh -Discharge: -20 ˚C up to +65 ˚C Maximum Discharge current: 30 mA cont. - Charge 0˚C up to +65 ˚C Key features: Good recovery characteristics after long storage period and deep *See cell datasheet for further details discharge Physical characteristics: Sealed rechargeable Ni-MH Button Cells from VARTA Microbattery can be Width: 15.5 mm operated in any position Height: 7.5 mm The cells complete the manufacturing process in a charged state Length: 36.5 mm Because of time and temperature depending self- discharge, the state of charge upon receipt can not be precisely defined. Typical weight: 9.0 g Before use, therefore, sealed Ni-MH cells should be recharged. Connector: Molex 51021-200 Wire length: 10.0 mm Main Applications: RTC-Backup Memory-Backup Sensor Network Supply Quality – Made in Germany Manufactured on highly automated lines Direct replacement for Ni-Cd No memory effect 0% lead, 0% mercury, and 0 % cadmium All cells have built in safety vent for security in abuse conditions UL recognition under file BBET2.MH13654 ISO 9000 certified for design and manufacture of rechargeable mass type cells and batteries. -

Unix Shell 1 Introduction 2 Description
CMSC 23000 Operating Systems Project 1 Winter 2006 January 6 Unix shell Due: January 20 1 Introduction Despite recent bad press, an unnamed agency of the government is eager to continue the fight against enemies of the state. To this aim, they have contracted with you to write a new Unix shell called the “Freedom shell” (or fsh). This shell will allow responsible unnamed agencies to monitor the activities of suspected terrorists, environmental activists, and other “bad actors.” Your task is to write an implementation of the fsh using POSIX threads (pthreads). The fsh is a simple Unix shell that allows a third party to remotely monitor shell commands. Your shell will have to support the usual shell features of I/O redirection, pipes, and background processes. But it also has to provide this clandestine monitoring feature. 2 Description The input to the fsh is a sequence of commands, each provided on a separate line of input text typed interactively at the keyboard: The fsh supports the following command syntax: Command ::= Program (| Command)opt | Program &opt | exit Program ::= Path Argsopt (< Path)opt (> Path)opt where a Path has the following syntax: Path ::= /opt Filename (/ Filename)∗ Filenames are non-empty sequences of letters, digits, or one of “.”, “-”, “+”, “=”, “@”, or “_.” 2.1 Signals Finally, your shell needs to ignore a single signal, SIGINT. This signal is generated when a user presses ctrl-C on the keyboard. When received, it should be passed to the currently running program, but it should not cause your shell to terminate. It should also have no effect on background jobs. -

Eaton VCP-W Vacuum Circuit Breakers
VCP-W Vacuum Circuit Breakers VCP-WG Vacuum Circuit Breakers for Export Generators Automotive Powering business worldwide Aerospace Eaton delivers the hundreds of power distribution products that are answering the demands of today’s fast changing world. We help our customers worldwide manage the power they need for buildings, aircraft, trucks, cars, machinery and entire businesses. And we do it in a way that consumes fewer resources. Next generation Powering Greener Buildings Truck transportation and Businesses Eaton is driving the Eaton’s Electrical Group is a development of new leading provider of power technologies – from hybrid quality, distribution and control drivetrains and emission control solutions that increase energy systems to advanced engine efficiency and improve power components – that reduce fuel quality, safety and reliability. consumption and emissions in Our solutions offer a growing trucks and cars. portfolio of “green” products and services, such as energy Higher expectations audits and real-time energy consumption monitoring. We continue to expand our Eaton’s Uninterruptible Power aerospace solutions and Hydraulics Supplies (UPS), variable-speed Hydraulics services to meet the needs of drives and lighting controls help new aviation platforms, conserve energy and increase including the high-flying light jet efficiency. and very light jet markets. Building on our strengths Our hydraulics business combines localised service and support with an innovative portfolio of fluid power solutions to answer the needs of global -

Rc the Plan 9 Shell
Rc ߞ The Plan 9 Shell Tom Duff [email protected]−labs.com ABSTRACT Rc is a command interpreter for Plan 9 that provides similar facilities to UNIXߣs Bourne shell, with some small additions and less idiosyncratic syntax. This paper uses numerous examples to describe rcߣs features, and contrasts rc with the Bourne shell, a model that many readers will be familiar with. 1. Introduction Rc is similar in spirit but different in detail from UNIXߣs Bourne shell. This paper describes rcߣs principal features with many small examples and a few larger ones. It assumes familiarity with the Bourne shell. 2. Simple commands For the simplest uses rc has syntax familiar to Bourne-shell users. All of the fol lowing behave as expected: date cat /lib/news/build who >user.names who >>user.names wc <file echo [a−f]*.c who | wc who; date vc *.c & mk && v.out /*/bin/fb/* rm −r junk || echo rm failed! 3. Quotation An argument that contains a space or one of rcߣs other syntax characters must be enclosed in apostrophes (’): rm ’odd file name’ An apostrophe in a quoted argument must be doubled: echo ’How’’s your father?’ 4. Patterns An unquoted argument that contains any of the characters *?[is a pattern to be matched against file names. A * character matches any sequence of characters, ? matches any single character, and [class] matches any character in the class, unless the first character of class is ~, in which case the class is complemented. The class may 2 also contain pairs of characters separated by −, standing for all characters lexically between the two.