Investigating Carbocations Using High Speed Ball Milling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Directive Effects in Abstraction Reactions of the Phenyl Radical Robert Frederick Bridger Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1963 Directive effects in abstraction reactions of the phenyl radical Robert Frederick Bridger Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Bridger, Robert Frederick, "Directive effects in abstraction reactions of the phenyl radical " (1963). Retrospective Theses and Dissertations. 2336. https://lib.dr.iastate.edu/rtd/2336 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been 63-5170 microfilmed exactly as received BRIDGER, Robert Frederick, 1934- DIRECTIVE EFFECTS IN ABSTRACTION RE ACTIONS OF THE PHENYL RADICAL. Iowa State University of Science and Technology Ph.D., 1963 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan DIRECTIVE EFFECTS IN ABSTRACTION REACTIONS OF THE PHENYL RADICAL by Robert Frederick Bridger A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Organic Chemistry Approved: Signature was redacted for privacy. In Charge of Major Work Signature was redacted for privacy. Head of Major Depart me6jb Signature was redacted for privacy. Iowa State University Of Science and Technology Ames, Iowa 1963 ii TABLE OF CONTENTS Page INTRODUCTION 1 LITERATURE REVIEW 3 RESULTS 6 DISCUSSION 36 EXPERIMENTAL 100 SUMMARY 149 REFERENCES CITED 151 ACKNOWLEDGEMENTS 158 iii LIST OF FIGURES Page Figure 1. -

Exam 1 (February 23, 2004) ID# ______
Chemistry 211 Name ___________________________ Exam 1 (February 23, 2004) ID# ___________________________ 10 1. You desire to synthesize 3-ethyl-3-pentanol starting with an ester. (i) What would be the name of the ester, and what is the name for the Grignard reagent (e.g., methyl magnesium bromide)? (ii) For the carbons shown in the product, show plausible hydrocarbons that you could start with to produce the ester and the Grignard reagent (as in a retrosynthesis). 12 2. (i) Show the step-by-step process required to produce propyllithium, which requires a free radical reaction mechanism, . (ii) Show the complete reaction mechanism for reaction between propyllithium and the correct ketone to produce 3-propyl-3-pentanol. (iii) Propose a possible reaction mechanism by which dipropyl cuprate (Cu+ with two propyl groups attached) could react with ethyl bromide to produce a new hydrocarbon. (This is a thinking exercise! So, think! () 8 3. As mentioned in the text, diethyl ether, pentane, and 1-butanol have similar molar masses, but different physical properties. Boiling points are 35oC, 36oC, and 117oC, respectively. Their respective solubilities in water are 7.5g/100mL, insoluble, and 9g/100mL. (i) Draw structures for each of these compounds. (ii) Justify the observed boiling points and their solubilities. 16 4. Draw structures of the following compounds 2,3-heptanediol isopropyllithium benzylmagnesium bromide benzoic acid benzaldehylde dimethyl sulfide t-butyl methanoate dibutyl ketone 12 5. Alcohols can be oxidized to produce other compounds, and can be produced by reduction. For the reactions shown below, show the structure for the expected product (if reaction does not occur, state: No Reaction) when treated with the indicated oxidizing or reducing agents. -

Course Material 2.Pdf
Reactive Intermediates Source: https://www.askiitians.com/iit-jee-chemistry/organic-chemistry/iupac- and-goc/reaction-intermediates/ Table of Content • Carbocations • Carbanions • Free Radicals • Carbenes • Arenium Ions • Benzynes Synthetic intermediate are stable products which are prepared, isolated and purified and subsequently used as starting materials in a synthetic sequence. Reactive intermediate, on the other hand, are short lived and their importance lies in the assignment of reaction mechanisms on the pathway from the starting substrate to stable products. These reactive intermediates are not isolated, but are detected by spectroscopic methods, or trapped chemically or their presence is confirmed by indirect evidence. • Carbocations Carbocations are the key intermediates in several reactions and particularly in nucleophilic substitution reactions. Structure of Carbocations : Generally, in the carbocations the positively charged carbon atom is bonded to three other atoms and has no nonbonding electrons. It is sp 2 hybridized with a planar structure and bond angles of about 120°. There is a + vacant unhybridized p orbital which in the case of CH 3 lies perpendicular to the plane of C—H bonds. Stability of Carbocations: There is an increase in carbocation stability with additional alkyl substitution. Thus one finds that addition of HX to three typical olefins decreases in the order (CH 3)2C=CH 2>CH 3—CH = CH 2 > CH 2 = CH 2. This is due to the relative stabilities of the carbocations formed in the rate determining step which in turn follows from the fact that the stability is increased by the electron releasing methyl group (+I), three such groups being more effective than two, and two more effective than one. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

Lecture Outline a Closer Look at Carbocation Stabilities and SN1/E1 Reaction Rates We Learned That an Important Factor in the SN
Lecture outline A closer look at carbocation stabilities and SN1/E1 reaction rates We learned that an important factor in the SN1/E1 reaction pathway is the stability of the carbocation intermediate. The more stable the carbocation, the faster the reaction. But how do we know anything about the stabilities of carbocations? After all, they are very unstable, high-energy species that can't be observed directly under ordinary reaction conditions. In protic solvents, carbocations typically survive only for picoseconds (1 ps - 10–12s). Much of what we know (or assume we know) about carbocation stabilities comes from measurements of the rates of reactions that form them, like the ionization of an alkyl halide in a polar solvent. The key to connecting reaction rates (kinetics) with the stability of high-energy intermediates (thermodynamics) is the Hammond postulate. The Hammond postulate says that if two consecutive structures on a reaction coordinate are similar in energy, they are also similar in structure. For example, in the SN1/E1 reactions, we know that the carbocation intermediate is much higher in energy than the alkyl halide reactant, so the transition state for the ionization step must lie closer in energy to the carbocation than the alkyl halide. The Hammond postulate says that the transition state should therefore resemble the carbocation in structure. So factors that stabilize the carbocation also stabilize the transition state leading to it. Here are two examples of rxn coordinate diagrams for the ionization step that are reasonable and unreasonable according to the Hammond postulate. We use the Hammond postulate frequently in making assumptions about the nature of transition states and how reaction rates should be influenced by structural features in the reactants or products. -

Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and Its Interactions with Supramolecular Systems
View metadata,Downloaded citation and from similar orbit.dtu.dk papers on:at core.ac.uk May 03, 2019 brought to you by CORE provided by Online Research Database In Technology Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and its Interactions with Supramolecular Systems Reynisson, Johannes Publication date: 2000 Document Version Publisher's PDF, also known as Version of record Link back to DTU Orbit Citation (APA): Reynisson, J. (2000). Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and its Interactions with Supramolecular Systems. Roskilde: Risø National Laboratory. Denmark. Forskningscenter Risoe. Risoe-R, No. 1191(EN) General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. Users may download and print one copy of any publication from the public portal for the purpose of private study or research. You may not further distribute the material or use it for any profit-making activity or commercial gain You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Spectroscopic and Photophysical Properties of the Trioxatriangulenium Carbocation and its Interactions with Supramolecular Systems Jóhannes Reynisson OO O Risø National Laboratory, Roskilde, Denmark June 2000 1 Abstract Trioxatriangulenium (TOTA+, 4,8,12-trioxa-4,8,12,12c-tetrahydro- dibenzo[cd,mn]-pyrenylium) is a closed shell carbocation which is stable in its crystalline form and in polar solvents at ambient temperatures. -

Grignard Reaction: Synthesis of Triphenylmethanol
*NOTE: Grignard reactions are very moisture sensitive, so all the glassware in the reaction (excluding the work-up) should be dried in an oven with a temperature of > 100oC overnight. The following items require oven drying. They should be placed in a 150mL beaker, all labeled with a permanent marker. 1. 5mL conical vial (AKA: Distillation receiver). 2. Magnetic spin vane. 3. Claisen head. 4. Three Pasteur pipettes. 5. Two 1-dram vials (Caps EXCLUDED). 6. One 2-dram vial (Caps EXCLUDED). 7. Glass stirring rod 8. Adaptor (19/22.14/20) Grignard Reaction: Synthesis of Triphenylmethanol Pre-Lab: In the “equations” section, besides the main equations, also: 1) draw the equation for the production of the byproduct, Biphenyl. 2) what other byproduct might occur in the reaction? Why? In the “observation” section, draw data tables in the corresponding places, each with 2 columns -- one for “prediction” (by answering the following questions) and one for actual drops or observation. 1) How many drops of bromobenzene should you add? 2) How many drops of ether will you add to flask 2? 3) 100 µL is approximately how many drops? 4) What are the four signs of a chemical reaction? (Think back to Chem. 110) 5) How do the signs of a chemical reaction apply to this lab? The Grignard reaction is a useful synthetic procedure for forming new carbon- carbon bonds. This organometallic chemical reaction involves alkyl- or aryl-magnesium halides, known as Grignard 1 reagents. Grignard reagents are formed via the action of an alkyl or aryl halide on magnesium metal. -

Organic Chemistry Laboratory II Preparation of Triphenylmethanol (Grignard Reaction) Experiment Procedure (Printable Pdf Format)
grigdes http://www.organicchem.org/oc2web/lab/exp/grig/grigdesbenzophenone... Organic Chemistry II | Lecture | Laboratory Organic Chemistry Laboratory II Preparation of Triphenylmethanol (Grignard Reaction) Experiment Procedure (Printable pdf format) Introduction In this two-week experiment, triphenylmethanol will be synthesized through a Grignard reaction. Students will work individually to prepare the Grignard reagent by reacting bromobenzene with solid magnesium in diethylether. The prepared Grignard reagent, phenylmagnesium bromide, will then be combined with bezophenone to form the desired triphenylmethanol product. Procedure NOTE: All glassware must be extremely dry! Place glassware in the drying oven for at least 10 minutes before beginning the reaction. Preparation of the Grignard Reagent Add ~0.5g of magnesium turnings and a magnetic stir bar to a dry 100 ml round-bottomed flask, and place the flask in the drying oven for 30 minutes. Remove the flask from the oven, clamp it to a ring stand, insert a reflux condenser, fit the flask into a heating mantle and immediately attach a drying tube to the top of the reflux condenser. Position the flask in the heating mantle on a stirrer/hotplate. Set the reaction up on the side of the hood near to the faucet labeled for distillation. Allow the reaction flask to cool to room temperature completely before proceeding. While the flask is cooling, place a 150ml beaker into the drying oven. While the flask is cooling, add 3.5 g of dry bromobenzene to a dry 50 mL Erlenmeyer flask and dissolve it in 5.0 mL of anhydrous diethyl ether. Use a glass powder funnel to dispense the bromobenze and ether to the Erlenmyer flask. -

Haloalkanes and Haloarenes | 145
For more FREE DOWNLOADS, visit www.aspirationsinstitute.com Haloalkanes and Haloarenes | 145 UNIT 9 HALOALKANES AND HALOARENES Points to Remember 1. Haloalkanes (Alkyl halides) are halogen derivatives of alkanes with general formula [CnH2n + 1X]. (X = F, Cl, Br or I) 2. Haloarenes (Aryl halides) are halogen derivatives of arenes with general formula Ar – X. 3. Since halogen is more electronegative than C, hence C – X bond is polar. 4. Named Reactions : (a) Sandmeyer Reaction : (b) Finkelstein Reaction : R− X + NaI →dry acetone R−+ I NaX (X = Cl, Br) (c) Swartz Reaction : CH3 – Br + AgF → CH3 – F + AgBr Instead of Ag – F, other metallic fluoride like Hg2F2, CoF2 or SbF3 can also be used. (d) Wurtz Reaction : 2R−+ X 2Na→dry ether R−+ R 2NaX (e) Wurtz-Fittig Reaction : (f) Fittig Reaction : For more FREE DOWNLOADS, visit www.aspirationsinstitute.com 146 | Chemistry-XII 5. Nucleophilic Substitution Reactions : | – δ+ δ– – Nu + –C–X C–Nu + X | haloalkane 2 (a) Substitution nucleophilic bimolecular (SN ) : 3 1. 1º haloalkane 2. Bimolecular, 2nd order 3. One step Order of reactivity : 1º > 2º > 3º Deciding factor : Steric hindrance 1 (a) Substitution nucleophilic unimolecular (SN ) : slow CH3 – (CH )–C–Br + OH 33 step 1 C (CH )–C–OH (Nucleophile) 33 (r.d.s) CH CH 3 3 (Fast) Recemization Planar configuration 50% carbo cation relation (Racemic mixture is formed) & 50% inversion 1. 3º haloalkane 2. Unimolecular, 1st order 3. Two steps Order of reactivity : 3º > 2º > 1º Deciding factor : Stability of carbo cation ⊕ ⊕ * Allylic CH22= CH − C H and benzylic C H CH halides undergo 65 2 reaction via SN1 mechanism as the corresponding carbo cations are resonance stabilized. -

Working with Hazardous Chemicals
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

13C NMR Study of Co-Contamination of Clays with Carbon Tetrachloride
Environ. Sci. Technol. 1998, 32, 350-357 13 sometimes make it the equal of solid acids like zeolites or C NMR Study of Co-Contamination silica-aluminas. Benesi (7-9) measured the Hammett acidity of Clays with Carbon Tetrachloride function H0 for a number of clays; these H0 values range from +1.5 to -8.2 (in comparison to H0 )-12 for 100% ) and Benzene sulfuric acid and H0 5 for pure acetic acid). Therefore, one can expect that certain chemical transformations might occur in/on clays that are similar to what are observed in zeolite TING TAO AND GARY E. MACIEL* systems. Thus, it is of interest to examine what happens Department of Chemistry, Colorado State University, when carbon tetrachloride and benzene are ªco-contami- Fort Collins, Colorado 80523 nantsº in a clay. This kind of information would be useful in a long-term view for understanding chemical transforma- tions of contaminants in soil at contaminated sites. Data on 13 these phenomena could also be useful for designing predic- Both solid-sample and liquid-sample C NMR experiments tive models and/or effective pollution remediation strategies. have been carried out to identify the species produced by Solid-state NMR results, based on 13C detection and line the reaction between carbon tetrachloride and benzene narrowing by magic angle spinning (MAS) and high-power when adsorbed on clays, kaolinite, and montmorillonite. Liquid- 1H decoupling (10), have been reported on a variety of organic sample 13C and 1H NMR spectra of perdeuteriobenzene soil components such as humic samples (11-13). Appar- extracts confirm the identities determined by solid-sample ently, there have been few NMR studies concerned directly 13C NMR and provide quantitative measures of the amounts with elucidating the interactions of organic compounds with of the products identifiedsbenzoic acid, benzophenone, and soil or its major components. -
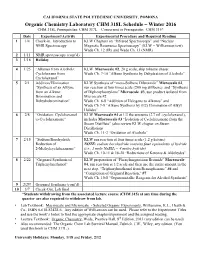
318 Lab Schedule
CALIFORNIA STATE POLYTECHNIC UNIVERSITY, POMONA Organic Chemistry Laboratory CHM 318L Schedule – Winter 2016 CHM 318L Prerequisites: CHM 317L Concurrent or Prerequisite: CHM 315* Date Experiment/Activity Experimental Procedure and Required Reading 1 1/4 Check-in. Introduction to KLW Chapters on “Infrared Spectroscopy” and “Nuclear NMR Spectroscopy Magnetic Resonance Spectroscopy” (KLW = Williamson text) Wade Ch. 12 (IR) and Wade Ch. 13 (NMR) 2 1/11 NMR spectroscopy (cont’d) 3 1/18 Holiday 4 1/25 “Alkenes from Alcohols: KLW Macroscale #2, 20 g scale, skip toluene chaser Cyclohexene from Wade Ch. 7-10 “Alkene Synthesis by Dehydration of Alcohols” Cyclohexanol” 5 2/1 Addition/Elimination KLW Synthesis of “meso-Stilbene Dibromide” Microscale #2, “Synthesis of an Alkyne run reaction at four times scale (200 mg stillbene); and “Synthesis from an Alkene: of Diphenylacetylene” Microscale #3, use product isolated from Bromination and Microscale #2 Dehydrobromination” Wade Ch. 8-8 “Addition of Halogens to Alkenes” and Wade Ch 7-9 “Alkene Synthesis by (E2) Elimination of Alkyl Halides” 6 2/8 “Oxidation: Cyclohexanol KLW Macroscale #4 at 1/3 the amounts (2.7 mL cyclohexanol), to Cyclohexanone” includes Macroscale #3 “Isolation of Cyclohexanone from the Steam Distillate” (also review KLW chapter on Steam Distillation) Wade Ch. 11-2 “Oxidation of Alcohols” 7 2/15 “Sodium Borohydride KLW run reaction at four times scale (1.2 g ketone) Reduction of NOTE: sodium borohydride contains four equivalents of hydride 2-Methylcyclohexanone” (i.e., 1 mole NaBH4 = 4 moles hydride) Wade Ch. 10-11 & 18-20 “Reductions of Ketones & Aldehydes” 8 2/22 “Grignard Synthesis of KLW preparation of “Phenylmagnesium Bromide” Macroscale Triphenylmethanol” #4, run reaction at 1/2 scale and then use the entire amount in the next step, “Triphenylmethanol from Benzophenone” #6 and “Completion of Grignard Reaction,” #7 Wade Ch.