Prediction and Classification of G Protein-Coupled Receptors Using Statistical and Machine Learning Algorithms
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Development and Maintenance of Epidermal Stem Cells in Skin Adnexa
International Journal of Molecular Sciences Review Development and Maintenance of Epidermal Stem Cells in Skin Adnexa Jaroslav Mokry * and Rishikaysh Pisal Medical Faculty, Charles University, 500 03 Hradec Kralove, Czech Republic; [email protected] * Correspondence: [email protected] Received: 30 October 2020; Accepted: 18 December 2020; Published: 20 December 2020 Abstract: The skin surface is modified by numerous appendages. These structures arise from epithelial stem cells (SCs) through the induction of epidermal placodes as a result of local signalling interplay with mesenchymal cells based on the Wnt–(Dkk4)–Eda–Shh cascade. Slight modifications of the cascade, with the participation of antagonistic signalling, decide whether multipotent epidermal SCs develop in interfollicular epidermis, scales, hair/feather follicles, nails or skin glands. This review describes the roles of epidermal SCs in the development of skin adnexa and interfollicular epidermis, as well as their maintenance. Each skin structure arises from distinct pools of epidermal SCs that are harboured in specific but different niches that control SC behaviour. Such relationships explain differences in marker and gene expression patterns between particular SC subsets. The activity of well-compartmentalized epidermal SCs is orchestrated with that of other skin cells not only along the hair cycle but also in the course of skin regeneration following injury. This review highlights several membrane markers, cytoplasmic proteins and transcription factors associated with epidermal SCs. Keywords: stem cell; epidermal placode; skin adnexa; signalling; hair pigmentation; markers; keratins 1. Epidermal Stem Cells as Units of Development 1.1. Development of the Epidermis and Placode Formation The embryonic skin at very early stages of development is covered by a surface ectoderm that is a precursor to the epidermis and its multiple derivatives. -

The G Protein-Coupled Receptor Subset of the Dog Genome Is More Similar
BMC Genomics BioMed Central Research article Open Access The G protein-coupled receptor subset of the dog genome is more similar to that in humans than rodents Tatjana Haitina1, Robert Fredriksson1, Steven M Foord2, Helgi B Schiöth*1 and David E Gloriam*2 Address: 1Department of Neuroscience, Functional Pharmacology, Uppsala University, BMC, Box 593, 751 24, Uppsala, Sweden and 2GlaxoSmithKline Pharmaceuticals, New Frontiers Science Park, 3rd Avenue, Harlow CM19 5AW, UK Email: Tatjana Haitina - [email protected]; Robert Fredriksson - [email protected]; Steven M Foord - [email protected]; Helgi B Schiöth* - [email protected]; David E Gloriam* - [email protected] * Corresponding authors Published: 15 January 2009 Received: 20 August 2008 Accepted: 15 January 2009 BMC Genomics 2009, 10:24 doi:10.1186/1471-2164-10-24 This article is available from: http://www.biomedcentral.com/1471-2164/10/24 © 2009 Haitina et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: The dog is an important model organism and it is considered to be closer to humans than rodents regarding metabolism and responses to drugs. The close relationship between humans and dogs over many centuries has lead to the diversity of the canine species, important genetic discoveries and an appreciation of the effects of old age in another species. The superfamily of G protein-coupled receptors (GPCRs) is one of the largest gene families in most mammals and the most exploited in terms of drug discovery. -

The Orphan Receptor GPR17 Is Unresponsive to Uracil Nucleotides and Cysteinyl Leukotrienes S
Supplemental material to this article can be found at: http://molpharm.aspetjournals.org/content/suppl/2017/03/02/mol.116.107904.DC1 1521-0111/91/5/518–532$25.00 https://doi.org/10.1124/mol.116.107904 MOLECULAR PHARMACOLOGY Mol Pharmacol 91:518–532, May 2017 Copyright ª 2017 by The American Society for Pharmacology and Experimental Therapeutics The Orphan Receptor GPR17 Is Unresponsive to Uracil Nucleotides and Cysteinyl Leukotrienes s Katharina Simon, Nicole Merten, Ralf Schröder, Stephanie Hennen, Philip Preis, Nina-Katharina Schmitt, Lucas Peters, Ramona Schrage,1 Celine Vermeiren, Michel Gillard, Klaus Mohr, Jesus Gomeza, and Evi Kostenis Molecular, Cellular and Pharmacobiology Section, Institute of Pharmaceutical Biology (K.S., N.M., Ral.S., S.H., P.P., N.-K.S, L.P., J.G., E.K.), Research Training Group 1873 (K.S., E.K.), Pharmacology and Toxicology Section, Institute of Pharmacy (Ram.S., K.M.), University of Bonn, Bonn, Germany; UCB Pharma, CNS Research, Braine l’Alleud, Belgium (C.V., M.G.). Downloaded from Received December 16, 2016; accepted March 1, 2017 ABSTRACT Pairing orphan G protein–coupled receptors (GPCRs) with their using eight distinct functional assay platforms based on label- cognate endogenous ligands is expected to have a major im- free pathway-unbiased biosensor technologies, as well as molpharm.aspetjournals.org pact on our understanding of GPCR biology. It follows that the canonical second-messenger or biochemical assays. Appraisal reproducibility of orphan receptor ligand pairs should be of of GPR17 activity can be accomplished with neither the coapplica- fundamental importance to guide meaningful investigations into tion of both ligand classes nor the exogenous transfection of partner the pharmacology and function of individual receptors. -

Edinburgh Research Explorer
Edinburgh Research Explorer International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list Citation for published version: Davenport, AP, Alexander, SPH, Sharman, JL, Pawson, AJ, Benson, HE, Monaghan, AE, Liew, WC, Mpamhanga, CP, Bonner, TI, Neubig, RR, Pin, JP, Spedding, M & Harmar, AJ 2013, 'International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands', Pharmacological reviews, vol. 65, no. 3, pp. 967-86. https://doi.org/10.1124/pr.112.007179 Digital Object Identifier (DOI): 10.1124/pr.112.007179 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Pharmacological reviews Publisher Rights Statement: U.S. Government work not protected by U.S. copyright General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 1521-0081/65/3/967–986$25.00 http://dx.doi.org/10.1124/pr.112.007179 PHARMACOLOGICAL REVIEWS Pharmacol Rev 65:967–986, July 2013 U.S. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Profiling G Protein-Coupled Receptors of Fasciola Hepatica Identifies Orphan Rhodopsins Unique to Phylum Platyhelminthes
bioRxiv preprint doi: https://doi.org/10.1101/207316; this version posted October 23, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Profiling G protein-coupled receptors of Fasciola hepatica 2 identifies orphan rhodopsins unique to phylum 3 Platyhelminthes 4 5 Short title: Profiling G protein-coupled receptors (GPCRs) in Fasciola hepatica 6 7 Paul McVeigh1*, Erin McCammick1, Paul McCusker1, Duncan Wells1, Jane 8 Hodgkinson2, Steve Paterson3, Angela Mousley1, Nikki J. Marks1, Aaron G. Maule1 9 10 11 1Parasitology & Pathogen Biology, The Institute for Global Food Security, School of 12 Biological Sciences, Queen’s University Belfast, Medical Biology Centre, 97 Lisburn 13 Road, Belfast, BT9 7BL, UK 14 15 2 Institute of Infection and Global Health, University of Liverpool, Liverpool, UK 16 17 3 Institute of Integrative Biology, University of Liverpool, Liverpool, UK 18 19 * Corresponding author 20 Email: [email protected] 21 1 bioRxiv preprint doi: https://doi.org/10.1101/207316; this version posted October 23, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 22 Abstract 23 G protein-coupled receptors (GPCRs) are established drug targets. Despite their 24 considerable appeal as targets for next-generation anthelmintics, poor understanding 25 of their diversity and function in parasitic helminths has thwarted progress towards 26 GPCR-targeted anti-parasite drugs. -
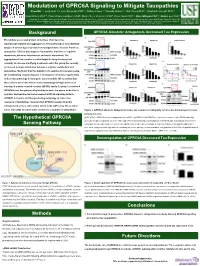
Background the Hypothetical GPRC6A Sensing Pathway
Modulation of GPRC6A Signaling to Mitigate Tauopathies Chao Ma1,2, Jerry Hunt1,3, Leslie Sandusky PhD1,3, William Fraser1,3, Ronald Alvarez1,3, Dali Zheng PhD1,4, Siddharth Kamath PhD1,2, Chad Dickey PhD1,4, Hans Bräuner-Osborne PhD5, Daniel Sejer Pedersen PhD5, Kevin Nash PhD1,2, Dave Morgan PhD1,2, Daniel Lee PhD1,3 1:Byrd Alzheimer‘s Institute, University of South Florida, Tampa, FL 33613, USA. 2:Morsani College of Medicine, Department of Molecular Pharmacology and Physiology, University of South Florida, Tampa, FL 33612, USA. 3:College of Pharmacy, Department of Pharmaceutical Sciences, University of South Florida, Tampa, FL 33612, USA. 4:Morsani College of Medicine, Department of Molecular Medicine, University of South Florida, Tampa, FL 33612, USA. 5:Department of Drug Design and Pharmacology, Faculty of Health and Medical Sciences, University of Copenhagen, Jagtvej 162, 2100 Copenhagen, Denmark Background GPRC6A Allosteric Antagonists Decreased Tau Expression Microtubule associated protein named tau, often becomes hyperphosphorylated and aggregates to form pathological neurofibrillary tangles in several age-associated neurodegenerative diseases known as tauopathies. Clinical phenotypes of tauopathies manifest as cognitive impairment, behavior disturbances and motor impairment. The aggregation of tau remains a central target for drug discovery, but currently no disease-modifying treatments exist. Our group has recently uncovered a unique interaction between L-arginine metabolism and tauopathies. We found that the depletion of L-arginine by overexpressing the metabolizing enzyme arginase 1 and arginine deiminase significantly reduced tau pathology in transgenic mouse models. We speculate that these effects associate with increased autophagy through amino acid sensing. G protein-coupled receptor (GPCR), family C, group 6, member A (GPRC6A) was deorphanized by binding to basic L-α amino acids, like L- arginine. -

Supplementary Table S5. Differentially Expressed Gene Lists of PD-1High CD39+ CD8 Tils According to 4-1BB Expression Compared to PD-1+ CD39- CD8 Tils
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Immunother Cancer Supplementary Table S5. Differentially expressed gene lists of PD-1high CD39+ CD8 TILs according to 4-1BB expression compared to PD-1+ CD39- CD8 TILs Up- or down- regulated genes in Up- or down- regulated genes Up- or down- regulated genes only PD-1high CD39+ CD8 TILs only in 4-1BBneg PD-1high CD39+ in 4-1BBpos PD-1high CD39+ CD8 compared to PD-1+ CD39- CD8 CD8 TILs compared to PD-1+ TILs compared to PD-1+ CD39- TILs CD39- CD8 TILs CD8 TILs IL7R KLRG1 TNFSF4 ENTPD1 DHRS3 LEF1 ITGA5 MKI67 PZP KLF3 RYR2 SIK1B ANK3 LYST PPP1R3B ETV1 ADAM28 H2AC13 CCR7 GFOD1 RASGRP2 ITGAX MAST4 RAD51AP1 MYO1E CLCF1 NEBL S1PR5 VCL MPP7 MS4A6A PHLDB1 GFPT2 TNF RPL3 SPRY4 VCAM1 B4GALT5 TIPARP TNS3 PDCD1 POLQ AKAP5 IL6ST LY9 PLXND1 PLEKHA1 NEU1 DGKH SPRY2 PLEKHG3 IKZF4 MTX3 PARK7 ATP8B4 SYT11 PTGER4 SORL1 RAB11FIP5 BRCA1 MAP4K3 NCR1 CCR4 S1PR1 PDE8A IFIT2 EPHA4 ARHGEF12 PAICS PELI2 LAT2 GPRASP1 TTN RPLP0 IL4I1 AUTS2 RPS3 CDCA3 NHS LONRF2 CDC42EP3 SLCO3A1 RRM2 ADAMTSL4 INPP5F ARHGAP31 ESCO2 ADRB2 CSF1 WDHD1 GOLIM4 CDK5RAP1 CD69 GLUL HJURP SHC4 GNLY TTC9 HELLS DPP4 IL23A PITPNC1 TOX ARHGEF9 EXO1 SLC4A4 CKAP4 CARMIL3 NHSL2 DZIP3 GINS1 FUT8 UBASH3B CDCA5 PDE7B SOGA1 CDC45 NR3C2 TRIB1 KIF14 TRAF5 LIMS1 PPP1R2C TNFRSF9 KLRC2 POLA1 CD80 ATP10D CDCA8 SETD7 IER2 PATL2 CCDC141 CD84 HSPA6 CYB561 MPHOSPH9 CLSPN KLRC1 PTMS SCML4 ZBTB10 CCL3 CA5B PIP5K1B WNT9A CCNH GEM IL18RAP GGH SARDH B3GNT7 C13orf46 SBF2 IKZF3 ZMAT1 TCF7 NECTIN1 H3C7 FOS PAG1 HECA SLC4A10 SLC35G2 PER1 P2RY1 NFKBIA WDR76 PLAUR KDM1A H1-5 TSHZ2 FAM102B HMMR GPR132 CCRL2 PARP8 A2M ST8SIA1 NUF2 IL5RA RBPMS UBE2T USP53 EEF1A1 PLAC8 LGR6 TMEM123 NEK2 SNAP47 PTGIS SH2B3 P2RY8 S100PBP PLEKHA7 CLNK CRIM1 MGAT5 YBX3 TP53INP1 DTL CFH FEZ1 MYB FRMD4B TSPAN5 STIL ITGA2 GOLGA6L10 MYBL2 AHI1 CAND2 GZMB RBPJ PELI1 HSPA1B KCNK5 GOLGA6L9 TICRR TPRG1 UBE2C AURKA Leem G, et al. -

Flow Reagents Single Color Antibodies CD Chart
CD CHART CD N° Alternative Name CD N° Alternative Name CD N° Alternative Name Beckman Coulter Clone Beckman Coulter Clone Beckman Coulter Clone T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells T Cells B Cells Granulocytes NK Cells Macrophages/Monocytes Platelets Erythrocytes Stem Cells Dendritic Cells Endothelial Cells Epithelial Cells CD1a T6, R4, HTA1 Act p n n p n n S l CD99 MIC2 gene product, E2 p p p CD223 LAG-3 (Lymphocyte activation gene 3) Act n Act p n CD1b R1 Act p n n p n n S CD99R restricted CD99 p p CD224 GGT (γ-glutamyl transferase) p p p p p p CD1c R7, M241 Act S n n p n n S l CD100 SEMA4D (semaphorin 4D) p Low p p p n n CD225 Leu13, interferon induced transmembrane protein 1 (IFITM1). p p p p p CD1d R3 Act S n n Low n n S Intest CD101 V7, P126 Act n p n p n n p CD226 DNAM-1, PTA-1 Act n Act Act Act n p n CD1e R2 n n n n S CD102 ICAM-2 (intercellular adhesion molecule-2) p p n p Folli p CD227 MUC1, mucin 1, episialin, PUM, PEM, EMA, DF3, H23 Act p CD2 T11; Tp50; sheep red blood cell (SRBC) receptor; LFA-2 p S n p n n l CD103 HML-1 (human mucosal lymphocytes antigen 1), integrin aE chain S n n n n n n n l CD228 Melanotransferrin (MT), p97 p p CD3 T3, CD3 complex p n n n n n n n n n l CD104 integrin b4 chain; TSP-1180 n n n n n n n p p CD229 Ly9, T-lymphocyte surface antigen p p n p n -

In-Depth Characterization of the Wnt-Signaling/Β-Catenin Pathway In
Götzel et al. BMC Gastroenterology (2019) 19:38 https://doi.org/10.1186/s12876-019-0957-5 RESEARCH ARTICLE Open Access In-depth characterization of the Wnt- signaling/β-catenin pathway in an in vitro model of Barrett’s sequence Katharina Götzel1, Olga Chemnitzer1, Luisa Maurer1, Arne Dietrich1,2, Uwe Eichfeld1, Orestis Lyros1, Yusef Moulla1, Stefan Niebisch1, Matthias Mehdorn1, Boris Jansen-Winkeln1, Michael Vieth3, Albrecht Hoffmeister4, Ines Gockel1 and René Thieme1* Abstract Background: An altered Wnt-signaling activation has been reported during Barrett’s esophagus progression, but with rarely detected mutations in APC and β-catenin (CTNNB1) genes. Methods: In this study, a robust in-depth expression pattern analysis of frizzled receptors, co-receptors, the Wnt- ligands Wnt3a and Wnt5a, the Wnt-signaling downstream targets Axin2, and CyclinD1, as well as the activation of the intracellular signaling kinases Akt and GSK3β was performed in an in vitro cell culture model of Barrett’s esophagus. Representing the Barrett’s sequence, we used normal esophageal squamous epithelium (EPC-1, EPC-2), metaplasia (CP-A) and dysplasia (CP-B) to esophageal adenocarcinoma (EAC) cell lines (OE33, OE19) and primary specimens of squamous epithelium, metaplasia and EAC. Results: A loss of Wnt3a expression was observed beginning from the metaplastic cell line CP-A towards dysplasia (CP-B) and EAC (OE33 and OE19), confirmed by a lower staining index of WNT3A in Barrett’s metaplasia and EAC, than in squamous epithelium specimens. Frizzled 1–10 expression analysis revealed a distinct expression pattern, showing the highest expression for Fzd2, Fzd3, Fzd4, Fzd5, Fzd7, and the co-receptor LRP5/6 in EAC cells, while Fzd3 and Fzd7 were rarely expressed in primary specimens from squamous epithelium. -

ADGRE1 (EMR1, F4/80) Is a Rapidly-Evolving Gene Expressed In
Edinburgh Research Explorer ADGRE1 (EMR1, F4/80) Is a Rapidly-Evolving Gene Expressed in Mammalian Monocyte-Macrophages Citation for published version: Waddell, L, Lefevre, L, Bush, S, Raper, A, Young, R, Lisowski, Z, McCulloch, M, Muriuki, C, Sauter, K, Clark, E, Irvine, KN, Pridans, C, Hope, J & Hume, D 2018, 'ADGRE1 (EMR1, F4/80) Is a Rapidly-Evolving Gene Expressed in Mammalian Monocyte-Macrophages' Frontiers in Immunology, vol. 9, 2246. DOI: 10.3389/fimmu.2018.02246 Digital Object Identifier (DOI): 10.3389/fimmu.2018.02246 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Frontiers in Immunology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 05. Apr. 2019 ORIGINAL RESEARCH published: 01 October 2018 doi: 10.3389/fimmu.2018.02246 ADGRE1 (EMR1, F4/80) Is a Rapidly-Evolving Gene Expressed in Mammalian Monocyte-Macrophages Lindsey A. Waddell 1†, Lucas Lefevre 1†, Stephen J. Bush 2†, Anna Raper 1, Rachel Young 1, Zofia M. -

Endothelial Loss of Fzd5 Stimulates PKC/Ets1-Mediated Transcription of Angpt2 and Flt1
Angiogenesis https://doi.org/10.1007/s10456-018-9625-6 ORIGINAL PAPER Endothelial loss of Fzd5 stimulates PKC/Ets1-mediated transcription of Angpt2 and Flt1 Maarten M. Brandt1 · Christian G. M. van Dijk2 · Ihsan Chrifi1 · Heleen M. Kool3 · Petra E. Bürgisser3 · Laura Louzao‑Martinez2 · Jiayi Pei2 · Robbert J. Rottier3 · Marianne C. Verhaar2 · Dirk J. Duncker1 · Caroline Cheng1,2 Received: 19 January 2018 / Accepted: 22 May 2018 © The Author(s) 2018 Abstract Aims Formation of a functional vascular system is essential and its formation is a highly regulated process initiated during embryogenesis, which continues to play important roles throughout life in both health and disease. In previous studies, Fzd5 was shown to be critically involved in this process and here we investigated the molecular mechanism by which endothelial loss of this receptor attenuates angiogenesis. Methods and results Using short interference RNA-mediated loss-of-function assays, the function and mechanism of sign- aling via Fzd5 was studied in human endothelial cells (ECs). Our findings indicate that Fzd5 signaling promotes neoves- sel formation in vitro in a collagen matrix-based 3D co-culture of primary vascular cells. Silencing of Fzd5 reduced EC proliferation, as a result of G 0/G1 cell cycle arrest, and decreased cell migration. Furthermore, Fzd5 knockdown resulted in enhanced expression of the factors Angpt2 and Flt1, which are mainly known for their destabilizing effects on the vasculature. In Fzd5-silenced ECs, Angpt2 and Flt1 upregulation was induced by enhanced PKC signaling, without the involvement of canonical Wnt signaling, non-canonical Wnt/Ca2+-mediated activation of NFAT, and non-canonical Wnt/PCP-mediated activation of JNK.