Phase Behavior Prediction of Ternary Polymer Mixtures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lecture 15: 11.02.05 Phase Changes and Phase Diagrams of Single- Component Materials
3.012 Fundamentals of Materials Science Fall 2005 Lecture 15: 11.02.05 Phase changes and phase diagrams of single- component materials Figure removed for copyright reasons. Source: Abstract of Wang, Xiaofei, Sandro Scandolo, and Roberto Car. "Carbon Phase Diagram from Ab Initio Molecular Dynamics." Physical Review Letters 95 (2005): 185701. Today: LAST TIME .........................................................................................................................................................................................2� BEHAVIOR OF THE CHEMICAL POTENTIAL/MOLAR FREE ENERGY IN SINGLE-COMPONENT MATERIALS........................................4� The free energy at phase transitions...........................................................................................................................................4� PHASES AND PHASE DIAGRAMS SINGLE-COMPONENT MATERIALS .................................................................................................6� Phases of single-component materials .......................................................................................................................................6� Phase diagrams of single-component materials ........................................................................................................................6� The Gibbs Phase Rule..................................................................................................................................................................7� Constraints on the shape of -

Phase Diagrams
Module-07 Phase Diagrams Contents 1) Equilibrium phase diagrams, Particle strengthening by precipitation and precipitation reactions 2) Kinetics of nucleation and growth 3) The iron-carbon system, phase transformations 4) Transformation rate effects and TTT diagrams, Microstructure and property changes in iron- carbon system Mixtures – Solutions – Phases Almost all materials have more than one phase in them. Thus engineering materials attain their special properties. Macroscopic basic unit of a material is called component. It refers to a independent chemical species. The components of a system may be elements, ions or compounds. A phase can be defined as a homogeneous portion of a system that has uniform physical and chemical characteristics i.e. it is a physically distinct from other phases, chemically homogeneous and mechanically separable portion of a system. A component can exist in many phases. E.g.: Water exists as ice, liquid water, and water vapor. Carbon exists as graphite and diamond. Mixtures – Solutions – Phases (contd…) When two phases are present in a system, it is not necessary that there be a difference in both physical and chemical properties; a disparity in one or the other set of properties is sufficient. A solution (liquid or solid) is phase with more than one component; a mixture is a material with more than one phase. Solute (minor component of two in a solution) does not change the structural pattern of the solvent, and the composition of any solution can be varied. In mixtures, there are different phases, each with its own atomic arrangement. It is possible to have a mixture of two different solutions! Gibbs phase rule In a system under a set of conditions, number of phases (P) exist can be related to the number of components (C) and degrees of freedom (F) by Gibbs phase rule. -

Phase Transitions in Multicomponent Systems
Physics 127b: Statistical Mechanics Phase Transitions in Multicomponent Systems The Gibbs Phase Rule Consider a system with n components (different types of molecules) with r phases in equilibrium. The state of each phase is defined by P,T and then (n − 1) concentration variables in each phase. The phase equilibrium at given P,T is defined by the equality of n chemical potentials between the r phases. Thus there are n(r − 1) constraints on (n − 1)r + 2 variables. This gives the Gibbs phase rule for the number of degrees of freedom f f = 2 + n − r A Simple Model of a Binary Mixture Consider a condensed phase (liquid or solid). As an estimate of the coordination number (number of nearest neighbors) think of a cubic arrangement in d dimensions giving a coordination number 2d. Suppose there are a total of N molecules, with fraction xB of type B and xA = 1 − xB of type A. In the mixture we assume a completely random arrangement of A and B. We just consider “bond” contributions to the internal energy U, given by εAA for A − A nearest neighbors, εBB for B − B nearest neighbors, and εAB for A − B nearest neighbors. We neglect other contributions to the internal energy (or suppose them unchanged between phases, etc.). Simple counting gives the internal energy of the mixture 2 2 U = Nd(xAεAA + 2xAxBεAB + xBεBB) = Nd{εAA(1 − xB) + εBBxB + [εAB − (εAA + εBB)/2]2xB(1 − xB)} The first two terms in the second expression are just the internal energy of the unmixed A and B, and so the second term, depending on εmix = εAB − (εAA + εBB)/2 can be though of as the energy of mixing. -

Introduction to Phase Diagrams*
ASM Handbook, Volume 3, Alloy Phase Diagrams Copyright # 2016 ASM InternationalW H. Okamoto, M.E. Schlesinger and E.M. Mueller, editors All rights reserved asminternational.org Introduction to Phase Diagrams* IN MATERIALS SCIENCE, a phase is a a system with varying composition of two com- Nevertheless, phase diagrams are instrumental physically homogeneous state of matter with a ponents. While other extensive and intensive in predicting phase transformations and their given chemical composition and arrangement properties influence the phase structure, materi- resulting microstructures. True equilibrium is, of atoms. The simplest examples are the three als scientists typically hold these properties con- of course, rarely attained by metals and alloys states of matter (solid, liquid, or gas) of a pure stant for practical ease of use and interpretation. in the course of ordinary manufacture and appli- element. The solid, liquid, and gas states of a Phase diagrams are usually constructed with a cation. Rates of heating and cooling are usually pure element obviously have the same chemical constant pressure of one atmosphere. too fast, times of heat treatment too short, and composition, but each phase is obviously distinct Phase diagrams are useful graphical representa- phase changes too sluggish for the ultimate equi- physically due to differences in the bonding and tions that show the phases in equilibrium present librium state to be reached. However, any change arrangement of atoms. in the system at various specified compositions, that does occur must constitute an adjustment Some pure elements (such as iron and tita- temperatures, and pressures. It should be recog- toward equilibrium. Hence, the direction of nium) are also allotropic, which means that the nized that phase diagrams represent equilibrium change can be ascertained from the phase dia- crystal structure of the solid phase changes with conditions for an alloy, which means that very gram, and a wealth of experience is available to temperature and pressure. -

Phase Diagrams of Ternary -Conjugated Polymer Solutions For
polymers Article Phase Diagrams of Ternary π-Conjugated Polymer Solutions for Organic Photovoltaics Jung Yong Kim School of Chemical Engineering and Materials Science and Engineering, Jimma Institute of Technology, Jimma University, Post Office Box 378 Jimma, Ethiopia; [email protected] Abstract: Phase diagrams of ternary conjugated polymer solutions were constructed based on Flory-Huggins lattice theory with a constant interaction parameter. For this purpose, the poly(3- hexylthiophene-2,5-diyl) (P3HT) solution as a model system was investigated as a function of temperature, molecular weight (or chain length), solvent species, processing additives, and electron- accepting small molecules. Then, other high-performance conjugated polymers such as PTB7 and PffBT4T-2OD were also studied in the same vein of demixing processes. Herein, the liquid-liquid phase transition is processed through the nucleation and growth of the metastable phase or the spontaneous spinodal decomposition of the unstable phase. Resultantly, the versatile binodal, spinodal, tie line, and critical point were calculated depending on the Flory-Huggins interaction parameter as well as the relative molar volume of each component. These findings may pave the way to rationally understand the phase behavior of solvent-polymer-fullerene (or nonfullerene) systems at the interface of organic photovoltaics and molecular thermodynamics. Keywords: conjugated polymer; phase diagram; ternary; polymer solutions; polymer blends; Flory- Huggins theory; polymer solar cells; organic photovoltaics; organic electronics Citation: Kim, J.Y. Phase Diagrams of Ternary π-Conjugated Polymer 1. Introduction Solutions for Organic Photovoltaics. Polymers 2021, 13, 983. https:// Since Flory-Huggins lattice theory was conceived in 1942, it has been widely used be- doi.org/10.3390/polym13060983 cause of its capability of capturing the phase behavior of polymer solutions and blends [1–3]. -

Section 1 Introduction to Alloy Phase Diagrams
Copyright © 1992 ASM International® ASM Handbook, Volume 3: Alloy Phase Diagrams All rights reserved. Hugh Baker, editor, p 1.1-1.29 www.asminternational.org Section 1 Introduction to Alloy Phase Diagrams Hugh Baker, Editor ALLOY PHASE DIAGRAMS are useful to exhaust system). Phase diagrams also are con- terms "phase" and "phase field" is seldom made, metallurgists, materials engineers, and materials sulted when attacking service problems such as and all materials having the same phase name are scientists in four major areas: (1) development of pitting and intergranular corrosion, hydrogen referred to as the same phase. new alloys for specific applications, (2) fabrica- damage, and hot corrosion. Equilibrium. There are three types of equili- tion of these alloys into useful configurations, (3) In a majority of the more widely used commer- bria: stable, metastable, and unstable. These three design and control of heat treatment procedures cial alloys, the allowable composition range en- conditions are illustrated in a mechanical sense in for specific alloys that will produce the required compasses only a small portion of the relevant Fig. l. Stable equilibrium exists when the object mechanical, physical, and chemical properties, phase diagram. The nonequilibrium conditions is in its lowest energy condition; metastable equi- and (4) solving problems that arise with specific that are usually encountered inpractice, however, librium exists when additional energy must be alloys in their performance in commercial appli- necessitate the knowledge of a much greater por- introduced before the object can reach true stabil- cations, thus improving product predictability. In tion of the diagram. Therefore, a thorough under- ity; unstable equilibrium exists when no addi- all these areas, the use of phase diagrams allows standing of alloy phase diagrams in general and tional energy is needed before reaching meta- research, development, and production to be done their practical use will prove to be of great help stability or stability. -

Phase Diagrams a Phase Diagram Is Used to Show the Relationship Between Temperature, Pressure and State of Matter
Phase Diagrams A phase diagram is used to show the relationship between temperature, pressure and state of matter. Before moving ahead, let us review some vocabulary and particle diagrams. States of Matter Solid: rigid, has definite volume and definite shape Liquid: flows, has definite volume, but takes the shape of the container Gas: flows, no definite volume or shape, shape and volume are determined by container Plasma: atoms are separated into nuclei (neutrons and protons) and electrons, no definite volume or shape Changes of States of Matter Freezing start as a liquid, end as a solid, slowing particle motion, forming more intermolecular bonds Melting start as a solid, end as a liquid, increasing particle motion, break some intermolecular bonds Condensation start as a gas, end as a liquid, decreasing particle motion, form intermolecular bonds Evaporation/Boiling/Vaporization start as a liquid, end as a gas, increasing particle motion, break intermolecular bonds Sublimation Starts as a solid, ends as a gas, increases particle speed, breaks intermolecular bonds Deposition Starts as a gas, ends as a solid, decreases particle speed, forms intermolecular bonds http://phet.colorado.edu/en/simulation/states- of-matter The flat sections on the graph are the points where a phase change is occurring. Both states of matter are present at the same time. In the flat sections, heat is being removed by the formation of intermolecular bonds. The flat points are phase changes. The heat added to the system are being used to break intermolecular bonds. PHASE DIAGRAMS Phase diagrams are used to show when a specific substance will change its state of matter (alignment of particles and distance between particles). -

Acid Dissociation Constant - Wikipedia, the Free Encyclopedia Page 1
Acid dissociation constant - Wikipedia, the free encyclopedia Page 1 Help us provide free content to the world by donating today ! Acid dissociation constant From Wikipedia, the free encyclopedia An acid dissociation constant (aka acidity constant, acid-ionization constant) is an equilibrium constant for the dissociation of an acid. It is denoted by Ka. For an equilibrium between a generic acid, HA, and − its conjugate base, A , The weak acid acetic acid donates a proton to water in an equilibrium reaction to give the acetate ion and − + HA A + H the hydronium ion. Key: Hydrogen is white, oxygen is red, carbon is gray. Lines are chemical bonds. K is defined, subject to certain conditions, as a where [HA], [A−] and [H+] are equilibrium concentrations of the reactants. The term acid dissociation constant is also used for pKa, which is equal to −log 10 Ka. The term pKb is used in relation to bases, though pKb has faded from modern use due to the easy relationship available between the strength of an acid and the strength of its conjugate base. Though discussions of this topic typically assume water as the solvent, particularly at introductory levels, the Brønsted–Lowry acid-base theory is versatile enough that acidic behavior can now be characterized even in non-aqueous solutions. The value of pK indicates the strength of an acid: the larger the value the weaker the acid. In aqueous a solution, simple acids are partially dissociated to an appreciable extent in in the pH range pK ± 2. The a actual extent of the dissociation can be calculated if the acid concentration and pH are known. -
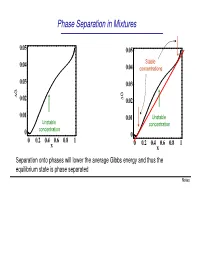
Phase Separation in Mixtures
Phase Separation in Mixtures 0.05 0.05 0.04 Stable 0.04 concentrations 0.03 0.03 G G ∆ 0.02 ∆ 0.02 0.01 0.01 Unstable Unstable concentration concentration 0 0 0 0.2 0.4 0.6 0.8 1 0 0.2 0.4 0.6 0.8 1 x x Separation onto phases will lower the average Gibbs energy and thus the equilibrium state is phase separated Notes Phase Separation versus Temperature Note that at higher temperatures the region of concentrations where phase separation takes place shrinks and G eventually disappears T increases ∆G = ∆U −T∆S This is because the term -T∆S becomes large at high temperatures. You can say that entropy “wins” over the potential energy cost at high temperatures Microscopically, the kinetic energy becomes x much larger than potential at high T, and the 1 x2 molecules randomly “run around” without noticing potential energy and thus intermix Oil and water mix at high temperature Notes Liquid-Gas Phase Separation in a Mixture A binary mixture can exist in liquid or gas phases. The liquid and gas phases have different Gibbs potentials as a function of mole fraction of one of the components of the mixture At high temperatures the Ggas <Gliquid because the entropy of gas is greater than that of liquid Tb2 T At lower temperatures, the two Gibbs b1 potentials intersect and separation onto gas and liquid takes place. Notes Boiling of a binary mixture Bubble point curve Dew point If we have a liquid at pint A that contains F A gas curve a molar fraction x1 of component 1 and T start heating it: E D - First the liquid heats to a temperature T’ C at constant x A and arrives to point B. -

Modeling of Phase Equilibria in Reactive Mixtures
Sumitomo Chemical Co., Ltd. Modeling of Phase Equilibria in Industrial Technology & Research Laboratory Reactive Mixtures Tetsuya SUZUTA Most chemical processes contain at least two of the three phases of solid, liquid and gas. Therefore it is essen- tial to fully understand phase equilibria for the logical design of processes and apparatus. The basic theory of phase equilibrium has already been established, but there are some exceptions which are not compliant with the theory. Many of those are influenced by chemical reactions within a certain phase or between different phases, so consideration of the reaction is necessary for precise reproduction of such unusual behavior. In this paper, some models for phase equilibria in reactive mixtures and several examples of their application in industrial plants are presented. This paper is translated from R&D Report, “SUMITOMO KAGAKU”, vol. 2018. Introduction1), 2) Modeling of Phase Equilibria Involving Chemical Reaction There are three states of matter: solid; liquid; and gas. Phase equilibrium is defined as a state in which 1. Chemical Reaction in Phase Equilibria matter and heat are balanced between at least two During a chemical reaction, a substance recombines phases and each phase is present stably. The phase- the atoms and atomic groups by itself or mutually with equilibrium relationship of the temperature, pressure other substances and generates a new substance. Some and the component composition is one of the impor- chemical reactions involve the cleavage or formation of tant properties in chemical processes. Numerous covalent bonding, and some occur due to the changes measured data of phase equilibrium had been accumu- in ion bonding or hydrogen bonding. -

CH 222 Oregon State University Week 6 Worksheet Notes
CH 222 Oregon State University Week 6 Worksheet Notes 1. Place the following compounds in order of decreasing strength of intermolecular forces. HF O2 CO2 HF > CO2 > O2 2. In liquid propanol, CH3CH2CH2OH, which intermolecular forces are present? Dispersion, hydrogen bonding and dipole-dipole forces are present. 3. Assign the appropriate labels to the phase diagram shown below. A) A = liquid, B = solid, C = gas, D = critical point B) A = gas, B = solid, C = liquid, D = triple point C) A = gas, B = liquid, C = solid, D = critical point D) A = solid, B = gas, C = liquid, D = supercritical fluid E) A = liquid, B = gas, C = solid, D = triple point 4. Consider the phase diagram below. If the dashed line at 1 atm of pressure is followed from 100 to 500°C, what phase changes will occur (in order of increasing temperature)? A) condensation, followed by vaporization B) sublimation, followed by deposition C) vaporization, followed by deposition D) fusion (melting), followed by vaporization E) No phase change will occur under the conditions specified. 5. Which of the following solutions will have the highest concentration of chloride ions? A) 0.10 M NaCl B) 0.10 M MgCl2 C) 0.10 M AlCl3 D) 0.05 M CaCl2 E) All of these solutions have the same concentration of chloride ions. 6. Commercial grade HCl solutions are typically 39.0% (by mass) HCl in water. Determine the molarity of the HCl, if the solution has a density of 1.20 g/mL. A) 7.79 M B) 10.7 M C) 12.8 M D) 9.35 M E) 13.9 M 7. -

Phase Diagrams
Phase diagrams 0.44 wt% of carbon in Fe microstructure of a lead–tin alloy of eutectic composition Primer Materials For Science Teaching Spring 2018 28.6.2018 What is a phase? A phase may be defined as a homogeneous portion of a system that has uniform physical and chemical characteristics Phase Equilibria • Equilibrium is a thermodynamic terms describes a situation in which the characteristics of the system do not change with time but persist indefinitely; that is, the system is stable • A system is at equilibrium if its free energy is at a minimum under some specified combination of temperature, pressure, and composition. The Gibbs Phase Rule This rule represents a criterion for the number of phases that will coexist within a system at equilibrium. Pmax = N + C C – # of components (material that is single phase; has specific stoichiometry; and has a defined melting/evaporation point) N – # of variable thermodynamic parameter (Temp, Pressure, Electric & Magnetic Field) Pmax – maximum # of phase(s) Gibbs Phase Rule – example Phase Diagram of Water C = 1 N = 1 (fixed pressure or fixed temperature) P = 2 fixed pressure (1atm) solid liquid vapor 0 0C 100 0C melting 1 atm. fixed pressure (0.0060373atm=611.73 Pa) boiling Ice Ih vapor 0.01 0C sublimation fixed temperature (0.01 oC) vapor liquid solid 611.73 Pa 109 Pa The Gibbs Phase Rule (2) This rule represents a criterion for the number of degree of freedom within a system at equilibrium. F = N + C - P F – # of degree of freedom: Temp, Pressure, composition (is the number of variables that can be changed independently without altering the phases that coexist at equilibrium) Example – Single Composition 1.