Osmium Tetroxide Original Commentary
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Study of the Modifications of Manganese Dioxide
U. S. Department of Commerce Research Paper RP1941 National Bureau of Standards Volume 41, December 1948 Part of the Journal of Research of the National Bureau of Standards Study of the Modifications of Mangan,ese Dioxide By Howard F. McMurdie and Esther Golovato P ast work on the modificat ions of manganese dioxide of in terest in dry-cell manufacture is revie wed. New X -ray data, at both room and elevaLcd temperatures, combined 'with differen tial hea ting curves lead to the concl usion that five type of manga nese dioxide exi t : (1) well-crystallized p yrolusite ; (2) gamma m a nganese diox ide, a poorly crystalli zed pyrol u site; (3) ramsdellite ; (4) cryp tomelane, a form co ntaining esse ntial p otassium or sodiulll.; a nd (5) delta m anganese dioxide, belie ved to be a poorly crystallize d cryptomela ne. The high-temperature X-r ay diffraction data indicated the phase cha nges t hat cause the heating curve effects. A new crystal fo rm of manganosic oxide (Mn30 4), stable above 1,1700 C, w ~ s fo und t o be cubic of spinel structure. Fincness dcten n inaLion by both thc nitrogen a dsorp t ion and Lhe X-ray line broadening me thods were made on selected samples. I. Introduction this equipment a fla t specinlen is used, and no During the years 1940- 46 there wa increased special teclmiq Ll es were employed to prcven t pre researeh on dry cells. This was stimulated by fcn 'cd orientation. It is r ealized that in a few increased demand for the cell as well a new u es cascs this may have res ulted in r elative intcnsitics for them , combined with certain hor Lages of raw that differ from those in other r eporLs. -

Sodium Periodate Solution (SP7469-G)
Page: 1/9 Safety Data Sheet according to OSHA HCS (29CFR 1910.1200) and WHMIS 2015 Regulations Revision: July 09, 2020 1 Identification · Product identifier · Trade name: Sodium Periodate Solution · Product code: SP7469-G · Recommended use and restriction on use · Recommended use: Laboratory chemicals · Restrictions on use: No relevant information available. · Details of the supplier of the Safety Data Sheet · Manufacturer/Supplier: AquaPhoenix Scientific, Inc. 860 Gitts Run Road Hanover, PA 17331 USA Tel +1 (717)632-1291 Toll-Free: (866)632-1291 [email protected] · Distributor: AquaPhoenix Scientific 860 Gitts Run Road, Hanover, PA 17331 (717) 632-1291 · Emergency telephone number: ChemTel Inc. (800)255-3924 (North America) +1 (813)248-0585 (International) 2 Hazard(s) identification · Classification of the substance or mixture Skin Irrit. 2 H315 Causes skin irritation. Eye Irrit. 2A H319 Causes serious eye irritation. STOT RE 1 H372 Causes damage to the thyroid through prolonged or repeated exposure. · Label elements · GHS label elements The product is classified and labeled according to the Globally Harmonized System (GHS). · Hazard pictograms: GHS07 GHS08 · Signal word: Danger · Hazard statements: H315 Causes skin irritation. H319 Causes serious eye irritation. H372 Causes damage to the thyroid through prolonged or repeated exposure. · Precautionary statements: P260 Do not breathe mist/vapors/spray. P264 Wash thoroughly after handling. (Cont'd. on page 2) 50.1.3 Page: 2/9 Safety Data Sheet according to OSHA HCS (29CFR 1910.1200) and WHMIS 2015 Regulations Revision: July 09, 2020 Trade name: Sodium Periodate Solution (Cont'd. of page 1) P270 Do not eat, drink or smoke when using this product. -

Production of Dialdehyde Cellulose and Periodate Regeneration: Towards Feasible Oxidation Processes
Production of Dialdehyde Cellulose and Periodate Regeneration: Towards feasible oxidation processes Produktion av dialdehydcellulosa och återgenerering av perjodat: Mot möjliga oxidationsprocesser Elisabeth Höglund Department of Engineering and Chemical Sciences Chemistry 30 hp Supervisors: Susanne Hansson, Stora Enso & Gunilla Carlsson, Karlstad University Examinator: Thomas Nilsson 2015-09-25 ABSTRACT Cellulose is an attractive raw material that has lately become more interesting thanks to its degradability and renewability and the environmental awareness of our society. With the intention to find new material properties and applications, studies on cellulose derivatization have increased. Dialdehyde cellulose (DAC) is a derivative that is produced by selective cleavage of the C2-C3 bond in an anhydroglucose unit in the cellulose chain, utilizing sodium periodate (NaIO4) that works as a strong oxidant. At a fixed temperature, the reaction time as well as the amount of added periodate affect the resulting aldehyde content. DAC has shown to have promising properties, and by disintegrating the dialdehyde fibers into fibrils, thin films with extraordinary oxygen barrier at high humidity can be achieved. Normally, barrier properties of polysccharide films deteriorate at higher humidity due to their hygroscopic character. This DAC barrier could therefore be a potential environmentally-friendly replacement for aluminum which is utilized in many food packages today. The aim of this study was to investigate the possibilities to produce dialdehyde cellulose at an industrial level, where the regeneration of consumed periodate plays a significant role to obtain a feasible process. A screening of the periodate oxidation of cellulose containing seven experiments was conducted by employing the program MODDE for experimental design. -

United States Patent (19) 11 Patent Number: 4,496,778 Myers Et Al
United States Patent (19) 11 Patent Number: 4,496,778 Myers et al. (45) Date of Patent: Jan. 29, 1985 (54) PROCESS FOR THE HYDROXYLATION OF 56 References Cited OLEFINS USING MOLECULAR OXYGEN, U.S. PATENT DOCUMENTS ANOSMIUM CONTAINING CATALYST, A COPPER CO-CATALYST, AND AN 2,773, 101 12/1956 Smith et al. ......................... 568/860 AROMATIC AMINE BASED PROMOTER 3,317,592 5/1967 Maclean et al. ... 568/860 3,337,635 8/1967 Norton et al. ....... 568/860 75 Inventors: Richard S. Myers, Fairlawn; Robert 4,390,739 6/1983 Michaelson et al. .... ..., 568/860 C. Michaelson, Waldwick; Richard FOREIGN PATENT DOCUMENTS G. Austin, Ridgewood, all of N.J. 32522 8/1974 Japan ................................... 568/860 73) Assignee: Exxon Research & Engineering Co., Primary Examiner-J. E. Evans Florham Park, N.J. Attorney, Agent, or Firm-Robert A. Maggio 21 Appl. No.: 538,190 57 ABSTRACT A process directed to the hydroxylation of olefins by 22 Filed: Oct. 3, 1983 reacting said olefins in the presence of oxygen, water, and a catalyst composition comprising (i) a catalytically 51 Int. Cl. ...................... C07C 29/04; CO7C 31/18; active osmium containing compound, (ii) a Co-catalyst C07C 31/22; CO7C 31/42 I comprising a copper containing compound such as 52 U.S.C. ................................. 568/860; 260/.397.2; CuBr2, and (iii) a Co-catalyst II capable of increasing 560/186; 562/587; 568/811; 568/821; 568/833; the rate and/or selectivity of the hydroxylation reac 568/838; 568/847 tion, such as pyridine is disclosed. 58 Field of Search .............. -

PATENT SPECIFICATION (11) 1 564 366 to (21) Application No
PATENT SPECIFICATION (11) 1 564 366 TO (21) Application No. 3795/77 (22) Filed 31 Jan. 1977 CO (31) Convention Application No. 661572 OO (32) Filed 26 Feb. 1976 in (33) United States of America (US) CD (44) Complete Specification published 10 April 1980 M5 (51) INT CL3 B01D 53/02 53/14 53/34 (52) Index at acceptance B1L 102 205 214 302 305 309 AF CIA SI86 S18Y S191 S19Y S420 S44Y S450 S451 S46Y S492 S493 SB G6R 1A10 (54) SALTS OF THE IODINE OXYACIDS IN THE IMPREGNATION OF ADSORBENT CHARCOAL FOR TRAPPING RADIOACTIVE METHYLIODIDE (71) We, UNITED STATES DEPARTMENT OF ENERGY, formerly United States Energy Research and Development Administration, Washington, District of Columbia 20545, United States of America, a duly constituted agency of the Government of the United States of America established by the Energy Reorganization 5 Act of 1974 (Public Law 93-438), do hereby declare the invention, for which we 5 pray that a patent may be granted to us and the method by which it is to be performed, to be particularly described in and by the following statement:— It is essential in nuclear power reactor operations to remove the radioiodine fission-product and the organic derivatives that are present in the reactor air cleaning 10 systems. This is done by passing the air stream through filters containing adsorbent 10 charcoal which is suitably impregnated with compounds capable of removing both elementary iodine and the organic iodide. The charcoal must remain at high efficiency during its long service time, often when confronted with adverse contaminants in the air. -

Potential Biocides: Iodine-Producing Pyrotechnics Full Paper
Full Paper 1 DOI: 10.1002/prep.201700037 2 3 4 Potential Biocides: Iodine-Producing Pyrotechnics 5 Jimmie C. Oxley,*[a] James L. Smith,[a] Matthew M. Porter,[a] Maxwell J. Yekel,[a] and Jeffrey A. Canaria[a] 6 7 8 9 Abstract: Currently there is a need for specialized py- measured with bomb calorimetry and extraction and analy- 10 rotechnic materials to combat the threat of biological sis of I2 by UV-Vis. Of the mixtures analyzed, calcium iodate 11 weapons. Materials have been characterized based on their and aluminum was found to be the highest producer of I2. 12 potential to produce heat and molecular iodine gas (I2)to The heat output of this mixture and others can be tuned by 13 kill spore-forming bacteria (e.g. anthrax). One formulation, adding more fuel, with the cost of some iodine. Products of 14 already proven to kill anthrax simulants, is diiodine pent- combustion were analyzed by thermal analysis (SDT), XPS, 15 oxide with aluminum; however, it suffers from poor stability XRD, and LC/MS. Evidence for various metal iodides and 16 and storage problems. The heat and iodine gas output from metal oxides was collected with these methods. 17 this mixture and candidate replacement mixtures were 18 Keywords: Keywords missing!!! 19 20 21 22 1 Introduction The pyrotechnic mixtures were mixed as dry loose pow- 23 ders using a Resodyne Lab Ram Acoustic Mixer (acceleration 24 Previously we examined a series of oxidizers and fuels to 35–40 G). Heat released from the ignition of the pyrotechnic 25 determine their potential as explosive threats [1]. -

Cellular Uptake and Toxicological Effects of Differently Sized Zinc Oxide Nanoparticles in Intestinal Cells †
toxics Article Cellular Uptake and Toxicological Effects of Differently Sized Zinc Oxide Nanoparticles in Intestinal Cells † Anna Mittag 1,* , Christian Hoera 2, Alexander Kämpfe 2 , Martin Westermann 3, Jochen Kuckelkorn 4, Thomas Schneider 1 and Michael Glei 1 1 Department of Nutritional Toxicology, Institute of Nutritional Sciences, Friedrich Schiller University Jena, Dornburger Straße 24, 07743 Jena, Germany; [email protected] (T.S.); [email protected] (M.G.) 2 German Environment Agency, Swimming Pool Water, Chemical Analytics, Heinrich-Heine-Straße 12, 08645 Bad Elster, Germany; [email protected] (C.H.); [email protected] (A.K.) 3 Electron Microscopy Centre, Friedrich Schiller University Jena, Ziegelmühlenweg 1, 07743 Jena, Germany; [email protected] 4 German Environment Agency, Toxicology of Drinking Water and Swimming Pool Water, Heinrich-Heine-Straße 12, 08645 Bad Elster, Germany; [email protected] * Correspondence: [email protected] † In respectful memory of Dr. Tamara Grummt. Abstract: Due to their beneficial properties, the use of zinc oxide nanoparticles (ZnO NP) is constantly increasing, especially in consumer-related areas, such as food packaging and food additives, which is leading to an increased oral uptake of ZnO NP. Consequently, the aim of our study was to investigate the cellular uptake of two differently sized ZnO NP (<50 nm and <100 nm; 12–1229 µmol/L) using two human intestinal cell lines (Caco-2 and LT97) and to examine the possible resulting toxic effects. ZnO NP (<50 nm and <100 nm) were internalized by both cell lines and led to intracellular changes. Citation: Mittag, A.; Hoera, C.; Kämpfe, A.; Westermann, M.; Both ZnO NP caused time- and dose-dependent cytotoxic effects, especially at concentrations of Kuckelkorn, J.; Schneider, T.; Glei, M. -

A Study of the Periodic Acid Oxidation of Cellulose Acetates of Low Acetyl
o A STUDY OF THE PERIODIC ACID OXIDATION OF CELLULOSE ACETATES OF LOW ACETYL CONTENT By Franklin Willard Herrick A THESIS Submitted to the School of Graduate Studies of Michigan State College of Agriculture and Applied Science in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Department of Chemistry 1950 ACOOTIBDGMENT Grateful recognition is given to Professor Bruce B. Hartsuch for his helpful guidance and inspiration throughout the course of this investigation. ********** ******** ****** **** ** * TABLE OF CONTENTS Page I INTRODUCTION.................................... ........ 1 The Structure of Cellulose. ..................... 1 The Present Problem.................................... 2 II GENERAL AMD HISTORICAL................................... 3 CELLULOSE ACETATE........................................ 3 PERIODATE OXIDATION OF CELLULOSE......................... 10 DISTRIBUTION OF HYDROXYL GROUPS IN CELLULOSE ACETATES.... 12 III EXPERIMENTAL............................................. 15 PREPARATION OF CELLULOSE ACETATE........................ 15 Materials.................................. .. ..... 15 Preparation of Standard Cellulose...................... 15 Preparation of Cellulose Acetates of Low Acetyl Content 16 Conditioning and Cutting of Standard Cellulose and Cellulose Acetate.................... 19 The Weighing of Linters ........................ 20 Analysis for Percentage of Combined Acetic Acid........ 21 Tabulation of Analyses of Cellulose Acetate Preparations 23 Calculation of the Degree -

The Two Faces of Titanium Dioxide Nanoparticles Bio-Camouflage in 3D
www.nature.com/scientificreports OPEN The two faces of titanium dioxide nanoparticles bio-camoufage in 3D bone spheroids Received: 23 October 2018 W. Souza1,2,3, S. G. Piperni3,4, P. Laviola1,3,5, A. L. Rossi4, Maria Isabel D. Rossi6, Accepted: 11 June 2019 Bráulio S. Archanjo7, P. E. Leite 1,2,8, M. H. Fernandes9,12, L. A. Rocha3,10, J. M. Granjeiro1,2,3,11 Published: xx xx xxxx & A. R. Ribeiro 2,3,5 Titanium (Ti) and its alloys are widely used in dental implants and hip-prostheses due to their excellent biocompatibility. Growing evidence support that surface degradation due to corrosion and wear processes, contribute to implant failure, since the release of metallic ions and wear particles generate local tissue reactions (peri-implant infammatory reactions). The generated ions and wear debris (particles at the micron and nanoscale) stay, in a frst moment, at the interface implant-bone. However, depending on their size, they can enter blood circulation possibly contributing to systemic reactions and toxicities. Most of the nanotoxicological studies with titanium dioxide nanoparticles (TiO2 NPs) use conventional two-dimensional cell culture monolayers to explore macrophage and monocyte activation, where limited information regarding bone cells is available. Recently three- dimensional models have been gaining prominence since they present a greater anatomical and physiological relevance. Taking this into consideration, in this work we developed a human osteoblast- like spheroid model, which closely mimics bone cell-cell interactions, providing a more realistic scenario for nanotoxicological studies. The treatment of spheroids with diferent concentrations of TiO2 NPs during 72 h did not change their viability signifcantly. -

United States Patent Office Attented Oct
3,536,479 United States Patent Office attented Oct. 27, 1970 2 a hydrogen pressure of at least about 15 pounds per 3,536,479 square inch gauge (p.S.i.g.). METHOD FOR THE PRODUCTION OF HIGH PURTY (OSMUM The concentrated osmium-containing solution can ad Alexander Ellis, Clarkson, Ontario, and Alan Manson, vantageously be prepared from a dilute osmium-contain Oakville, Ontario, Canada, assignors to The Interna 5 ing solution as described hereinafter. However, most gen tional Nicke Company, Inc., New York, N.Y., a cor erally the concentrated solution will be prepared by poration of Delaware scrubbing gases containing osmium tetroxide with a Solu No Drawing. Filled Dec. 13, 1967, Ser. No. 690,083 tion containing, by weight, about 5% to 40% sodium hy Claims priority, application Canada, Feb. 21, 1967, droxide to produce a sodium perosmate solution which 983,421 10 contains about 5 grams per liter (gp.l.) to about 100 nt. C. C22b 7/00, 11/04 g.p.l. of osmium, e.g., about 60 g.p.l. of osmium. Gen U.S. C. 75-108 29 Claims erally at this stage only minor amounts of ruthenium, i.e., less than about 0.5 g.p.l. of ruthenium, accompany the osmium and can be precipitated with only minor ABSTRACT OF THE DISCLOSURE 5 amounts of osmium occluded therein from the concen Metallic osmium is recovered from a slurry of an trated osmium-containing solution by adding a Water osmium-containing material to which sufficient hydro soluble, mild organic reducing agent such as methyl al chloric acid has been added to assure a final pH value cohol and ethyl alcohol. -

Modification of Manganese Dioxide Catalyst Owing to the Change in Gas Composition
Title Modification of Manganese Dioxide Catalyst Owing to the Change in Gas Composition Author(s) Kobayashi, Masayoshi; Kobayashi, Haruo Citation 北海道大學工學部研究報告, 69, 221-225 Issue Date 1973-11-15 Doc URL http://hdl.handle.net/2115/41166 Type bulletin (article) File Information 69_221-226.pdf Instructions for use Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP Modification of Manganese Dioxide Catalyst Owing to the Change in Gas Composition Masayoshi KoBAyAsm and Haruo KoBAyAsm (Received April 28, 1973) Abstruct Modification of the electrolytic manganese dioxide surface during oxidation of carbon monoxide was studied experimentaly by purteybing the reaction steady state with a stepwise change in the concentration of carbon monoxide. The transient behavior of the oxidation activity corresponded exactly to that of the amount of surface oxygen species having a higher oxidation power, OgL*, and moreover t};1e apparent first order rate constant predicted by the steady state kinetics varied in proportion to the amount of Oge’. lt was shown that the modification of catalyst surface owing to he change in gas composition affected the oxidation activity through the change of the amount of 03t* which was catalytically active for oxidation of cabon monoxide. 1. lntroductioR It has been pointed out’un3) that the original composition of soiid catalysts is modified as a result of the equi正ibration processes of the solid-gas system and the catalytic activity of the surface is thereby changed correspondiRg to the composition of the -
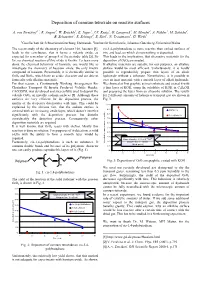
Deposition of Osmium Tetroxide on Reactive Surfaces
Deposition of osmium tetroxide on reactive surfaces A. von Zweidorf1,2, R. Angert1, W. Brüchle1, E. Jäger1, J.V. Kratz2, G. Langrock2, M. Mendel2, A. Nähler2, M. Schädel1, B. Schausten1, E. Schimpf1, E. Stiel1, N. Trautmann2, G. Wirth1 1Gesellschaft für Schwerionenforschung, Darmstadt, 2Institut für Kernchemie, Johannes Gutenberg-Universität Mainz The recent study of the chemistry of element 108, hassium [1], cis-1,4-polybutadiene is more reactive than etched surfaces of leads to the conclusion, that it forms a volatile oxide, as zinc and lead, on which almost nothing is deposited. expected for a member of group 8 of the periodic table [2]. So This leads to the implication, that alternative materials for the far, no chemical reaction of this oxide is known. To learn more deposition of OsO4 are needed. about the chemical behaviour of hassium, one would like to If alkaline materials are suitable for our purposes, an alkaline investigate the chemistry of hassium oxide, the only known surface would be most efficient. Unfortunately, it is hardly compound of hassium. Presumably, it is chemically similar to possible to reproducibly prepare thin layers of an alkali OsO4 and RuO4, which have an acidic character and are able to hydroxide without a substrate. Nevertheless, is it possible to form salts with alkaline materials. coat an inert material with a smooth layer of alkali hydroxide. For that reason, a Continuously Working Arrangement For We choosed at first graphite as inert substrate and coated it with Clusterless Transport Of In-situ Produced Volatile Oxides, a thin layer of KOH, using the solubility of KOH in C2H5OH CALLISTO, was developed and successfully used to deposit the and preparing the layer from an ethanolic solution.