Transformation of Hematopoietic Cells and Activation of JAK2-V617F by IL-27R, a Component of a Heterodimeric Type I Cytokine Receptor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

Mechanism of Homodimeric Cytokine Receptor Activation and Dysregulation by Oncogenic Mutations
This is a repository copy of Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations. White Rose Research Online URL for this paper: https://eprints.whiterose.ac.uk/155270/ Version: Accepted Version Article: Wilmes, Stephan, Hafer, Maximillian, Vuorio, Joni et al. (15 more authors) (2020) Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations. Science. pp. 643-652. ISSN 0036-8075 https://doi.org/10.1126/science.aaw3242 Reuse Items deposited in White Rose Research Online are protected by copyright, with all rights reserved unless indicated otherwise. They may be downloaded and/or printed for private study, or other acts as permitted by national copyright laws. The publisher or other rights holders may allow further reproduction and re-use of the full text version. This is indicated by the licence information on the White Rose Research Online record for the item. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ Submitted Manuscript: Confidential Title: Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations Authors: 5 Stephan Wilmes1, 2*, Maximillian Hafer1*, Joni Vuorio3,4, Julie A. Tucker5, Hauke Winkelmann1, Sara Löchte1, Tess A. Stanly5, Katiuska D. Pulgar Prieto5, Chetan Poojari3, Vivek Sharma3,6, Christian P. Richter1, Rainer Kurre1, Stevan R. Hubbard7, K. Christopher Garcia8,9, Ignacio Moraga2, Ilpo Vattulainen3,4,10†, Ian S. Hitchcock5† and Jacob Piehler1† Affiliations: 10 1 Department of Biology and Center of Cellular Nanoanalytics, University of Osnabrück, 49076 Osnabrück, Germany. -

Stony Brook University
SSStttooonnnyyy BBBrrrooooookkk UUUnnniiivvveeerrrsssiiitttyyy The official electronic file of this thesis or dissertation is maintained by the University Libraries on behalf of The Graduate School at Stony Brook University. ©©© AAAllllll RRRiiiggghhhtttsss RRReeessseeerrrvvveeeddd bbbyyy AAAuuuttthhhooorrr... Regulation of Dimerization and Activation of the Thrombopoietin Receptor A Dissertation Presented by Miki Itaya to The Graduate School in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biochemistry and Structural Biology Stony Brook University December 2012 Copyright by Miki Itaya 2012 Stony Brook University The Graduate School Miki Itaya We, the dissertation committee for the above candidate for the Doctor of Philosophy degree, hereby recommend acceptance of this dissertation. Steven O. Smith, Ph.D. - Dissertation Advisor Professor, Department of Biochemistry and Cell Biology Erwin London, Ph.D. - Chairperson of Defense Professor, Department of Biochemistry and Cell Biology Robert C. Rizzo, Ph.D. Associate Professor, Department of Applied Mathematics and Statistics Nancy Reich Marshall, Ph.D. Professor, Department of Molecular Genetics and Microbiology This dissertation is accepted by the Graduate School Charles Taber Interim Dean of the Graduate School ii Abstract of the Dissertation Regulation of Dimerization and Activation of the Thrombopoietin Receptor by Miki Itaya Doctor of Philosophy in Biochemistry and Structural Biology Stony Brook University 2012 The thrombopoietin receptor (TpoR) is -

Immuno-Endocrine Interactions in Intestinal Inflammation
IMMUNO-ENDOCRINE INTERACTIONS IN INTESTINAL INFLAMMATION PhD Thesis- Shajib, MS; McMaster University-Medical Sciences Immuno-endocrine interactions in intestinal inflammation By Md. Sharif Shajib, BSc. (Hons) A Thesis Submitted to the School of Graduate Studies in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy McMaster University © Copyright by Md. Sharif Shajib, 2017. PhD Thesis- Shajib, MS; McMaster University-Medical Sciences Descriptive notes Doctor of Philosophy (2017) McMaster University, Hamilton, Ontario (Medical Sciences) TITLE Immuno-endocrine interactions in intestinal inflammation AUTHOR Md. Sharif Shajib, BSc. (Hons) SUPERVISOR Dr. Waliul I. Khan NUMBER OF PAGES: XX, 292. II PhD Thesis- Shajib, MS; McMaster University-Medical Sciences Lay abstract The gut produces most of the serotonin found in our body, where it regulates many normal functions. A group of special cells, named enterochromaffin cells, produces nearly all of the serotonin in the gut. In diseases of the gut, especially ones that involve inflammation resulting in symptoms like abdominal pain, diarrhea and bleeding, the number of these cells and serotonin concentration are different from that in the normal gut. I found that these changes are controlled by a particular protein produced by immune cells, called interleukin-13, and alteration in serotonin levels, in turn, contributes to the inflammatory process. Our laboratory experiments with cells and animals establish this connection between interleukin-13 and serotonin in gut inflammation. We further confirm this association between interleukin-13 and serotonin in human inflammatory bowel disease. Moreover, we identify a potential genetic cause of these changes in serotonin concentrations which may ultimately result in inflammatory bowel disease. -
GM-CSF Receptor-Beta, Human, Recombinant Recombinant Human Granulocyte/Macrophage Colony Stimulating Factor Receptor Beta
GM-CSF Receptor-beta, human, recombinant Recombinant Human Granulocyte/Macrophage Colony Stimulating Factor Receptor beta Instruction Manual Catalog Number C-60430 Synonyms CSF2RB,Colony Stimulating Factor 2 Receptor, Beta, Low-Affinity (Granulocyte-Macrophage), GM-CSF/IL-3/IL-5 Receptor Common Beta Subunit, CDw131, IL3RB, SMDP5, IL5RB, Interleukin 3 Receptor/Granulocyte-Macrophage Colony Stimulating Factor 3 Receptor, Beta (High Affinity), Colony-Stimulating Factor-2 Receptor, Beta, Low-Affinity, GM-CSF/IL-3/IL-5 Receptor Common Beta-Chain, Cytokine Receptor Common Subunit Beta, CD131 Antigen, CD131 Description GM-CSF Receptor-beta, also known as CSF2RB is a member of the type I cytokine receptor family. CSF2RB is a high affinity receptor for interleukin-3, interleukin-5 as well as granulocyte- macrophage colony-stimulating factor. The CSF2RB unique form of receptor assembly applies also to IL-3 and IL-5 receptors, providing a structural basis for understanding their activation mechanism which is essential for the development of therapeutics. Human recombinant GM-CSF Receptor-beta produced in Sf9 cells is a single, glycosylated polypeptide chain of 435 amino acids (17-443 a.a), having a molecular mass of 49.7 kDa (migrates at 40-57 kDa on SDS-PAGE under reducing conditions). CSF2RB is fused to an 8 amino acid His-tag at the C-terminus and purified using proprietary chromatographic techniques. Quantity 10 µg Molecular Mass 49.7 kDa Source Sf9 insect cells Biological-Activity NA Specific Activity NA Formulation Sterile-filtered, clear protein solution (0.5 mg/ml) containing phosphate-buffered saline (pH 7.4) and 10% glycerol. Reconstitution Please Note: Always centrifuge product briefly before opening vial. -

Facteurs Génétiques De Prédisposition a La Maladie Coeliaque Et L’Oesophagite Éosinophilique
Université de Montréal FACTEURS GÉNÉTIQUES DE PRÉDISPOSITION A LA MALADIE COELIAQUE ET L’OESOPHAGITE ÉOSINOPHILIQUE Par Freha Nour el Hayet CHERIEF Département de Microbiologie et Immunologie Faculté de Médecine Mémoire présenté à la Faculté des études supérieures en vue de l’obtention du grade de Maîtrise en Microbiologie et Immunologie Novembre 2012 © Freha Nour el Hayet CHERIEF, 2012 i Université de Montréal Faculté des études supérieures Ce mémoire intitulé : FACTEURS GÉNÉTIQUES DE PRÉDISPOSITION A LA MALADIE COELIAQUE ET L’OESOPHAGITE ÉOSINOPHILIQUE Présenté par : Freha nour el hayet CHERIEF a été évalué par un jury composé des personnes suivantes : Ali Ahmad Président-rapporteur Idriss Djilali-Saïah Directeur de recherche Cristophe Faure Membre du jury ii Résumé Les maladies immunitaires chroniques incluant les maladies auto-immunes et inflammatoires touchent 20 à 25% de la population des pays occidentaux. La comparaison des taux de concordance chez les jumeaux ou l’histoire familiale de sujets atteints de la maladie cœliaque (maladie auto-immune de l’intestin) ou de l’œsophagite éosinophilique (maladie inflammatoire de l’œsophage) indiquent que des facteurs génétiques et environnementaux interviennent dans la susceptibilité à ces maladies. Cependant, ces études ne distinguent pas de manière claire la prédisposition génétique selon l’hétérogénéité clinique (enfants versus adultes) ou ethnique (stratification des populations). Méthodes. Les haplotypes HLA de prédisposition à la maladie cœliaque et les polymorphismes des gènes candidats -

WO 2018/067991 Al 12 April 2018 (12.04.2018) W !P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/067991 Al 12 April 2018 (12.04.2018) W !P O PCT (51) International Patent Classification: achusetts 021 15 (US). THE BROAD INSTITUTE, A61K 51/10 (2006.01) G01N 33/574 (2006.01) INC. [US/US]; 415 Main Street, Cambridge, Massachu C07K 14/705 (2006.01) A61K 47/68 (2017.01) setts 02142 (US). MASSACHUSETTS INSTITUTE OF G01N 33/53 (2006.01) TECHNOLOGY [US/US]; 77 Massachusetts Avenue, Cambridge, Massachusetts 02139 (US). (21) International Application Number: PCT/US2017/055625 (72) Inventors; and (71) Applicants: KUCHROO, Vijay K. [IN/US]; 30 Fairhaven (22) International Filing Date: Road, Newton, Massachusetts 02149 (US). ANDERSON, 06 October 2017 (06.10.2017) Ana Carrizosa [US/US]; 110 Cypress Street, Brookline, (25) Filing Language: English Massachusetts 02445 (US). MADI, Asaf [US/US]; c/o The Brigham and Women's Hospital, Inc., 75 Francis (26) Publication Language: English Street, Boston, Massachusetts 021 15 (US). CHIHARA, (30) Priority Data: Norio [US/US]; c/o The Brigham and Women's Hospital, 62/405,835 07 October 2016 (07.10.2016) US Inc., 75 Francis Street, Boston, Massachusetts 021 15 (US). REGEV, Aviv [US/US]; 15a Ellsworth Ave, Cambridge, (71) Applicants: THE BRIGHAM AND WOMEN'S HOSPI¬ Massachusetts 02139 (US). SINGER, Meromit [US/US]; TAL, INC. [US/US]; 75 Francis Street, Boston, Mass c/o The Broad Institute, Inc., 415 Main Street, Cambridge, (54) Title: MODULATION OF NOVEL IMMUNE CHECKPOINT TARGETS CD4 FIG. -

A Closer Look at JAK/STAT Signaling Pathway Emira Bousoik Chapman University
Chapman University Chapman University Digital Commons Pharmacy Faculty Articles and Research School of Pharmacy 7-31-2018 “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway Emira Bousoik Chapman University Hamidreza Montazeri Aliabadi Chapman University, [email protected] Follow this and additional works at: https://digitalcommons.chapman.edu/pharmacy_articles Part of the Amino Acids, Peptides, and Proteins Commons, Cancer Biology Commons, Cell Anatomy Commons, Cell Biology Commons, Enzymes and Coenzymes Commons, Oncology Commons, and the Other Pharmacy and Pharmaceutical Sciences Commons Recommended Citation Bousoik E, Montazeri Aliabadi H. “Do we know jack” about JAK? A closer look at JAK/STAT signaling pathway. Front. Oncol. 2018;8:287. doi: 10.3389/fonc.2018.00287 This Article is brought to you for free and open access by the School of Pharmacy at Chapman University Digital Commons. It has been accepted for inclusion in Pharmacy Faculty Articles and Research by an authorized administrator of Chapman University Digital Commons. For more information, please contact [email protected]. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway Comments This article was originally published in Frontiers in Oncology, volume 8, in 2018. DOI: 10.3389/ fonc.2018.00287 Creative Commons License This work is licensed under a Creative Commons Attribution 4.0 License. Copyright The uthora s This article is available at Chapman University Digital Commons: https://digitalcommons.chapman.edu/pharmacy_articles/590 -

Regulation of Cytokine Gene Expression in Immunity and Diseases Advances in Experimental Medicine and Biology
Advances in Experimental Medicine and Biology 941 Xiaojing Ma Editor Regulation of Cytokine Gene Expression in Immunity and Diseases Advances in Experimental Medicine and Biology Volume 941 Editorial Board IRUN R. COHEN, The Weizmann Institute of Science, Rehovot, Israel N.S. ABEL LAJTHA, Kline Institute for Psychiatric Research, Orangeburg, NY, USA JOHN D. LAMBRIS, University of Pennsylvania, Philadelphia, PA, USA RODOLFO PAOLETTI, University of Milan, Milan, Italy More information about this series at http://www.springer.com/series/5584 Xiaojing Ma Editor Regulation of Cytokine Gene Expression in Immunity and Diseases Editor Xiaojing Ma State Key Laboratory of Microbial Metabolism, Sheng Yushou Center of Cell Biology and Immunology, School of Life Science and Biotechnology Shanghai Jiaotong University, Shanghai , China Department of Microbiology and Immunology Weill Cornell Medical College NY , USA ISSN 0065-2598 ISSN 2214-8019 (electronic) Advances in Experimental Medicine and Biology ISBN 978-94-024-0919-2 ISBN 978-94-024-0921-5 (eBook) DOI 10.1007/978-94-024-0921-5 Library of Congress Control Number: 2016955715 © Springer Science+Business Media Dordrecht 2016 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifi cally the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfi lms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specifi c statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. -

Rabbit Anti-Phospho-IL2 Receptor Beta-SL11300R-FITC
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-phospho-IL2 Receptor beta SL11300R-FITC Product Name: Anti-phospho-IL2 Receptor beta (Tyr364)/FITC Chinese Name: FITC标记的磷酸化白介素2受体β链抗体 IL2 Receptor beta (phospho Y364); IL2 Receptor beta (phospho Tyr364); p-IL2 Receptor beta (Tyr364); Imterleukm-2 Receptor beta; CD 122; CD122; CD122 antigen; High affinity IL 2 receptor beta subunit; High affinity IL 2 receptor subunit beta; High affinity IL2 receptor beta subunit; High affinity IL2 receptor subunit beta; High affinity Alias: IL-2 receptor subunit beta; IL 2 receptor; IL-2 receptor; IL-2 receptor subunit beta; IL- 2R subunit beta; IL-2RB; IL15RB; IL2 R B; IL-2 R B; IL2 RB; IL2 receptor; IL2RB; Interleukin 2 receptor beta; Interleukin 2 receptor subunit beta; Interleukin-2 receptor subunit beta; OTTHUMP00000028799; P70 75; P7075; p75; IL2RB_HUMAN. Organism Species: Rabbit Clonality: Polyclonal React Species: Human,Mouse,Rat,Chicken,Pig,Cow,Horse,Sheep, ICC=1:50-200IF=1:50-200 Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 58kDawww.sunlongbiotech.com Cellular localization: The cell membrane Form: Lyophilized or Liquid Concentration: 1mg/ml KLH conjugated Synthesised phosphopeptide derived from human IL2 Receptor beta immunogen: around the phosphorylation site of Tyr364 Lsotype: IgG Purification: affinity purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. -

Generation Mechanisms of Soluble Cytokine Receptor
Mechanisms of Soluble Cytokine Receptor Generation Stewart J. Levine This information is current as J Immunol 2004; 173:5343-5348; ; of October 1, 2021. doi: 10.4049/jimmunol.173.9.5343 http://www.jimmunol.org/content/173/9/5343 References This article cites 113 articles, 70 of which you can access for free at: Downloaded from http://www.jimmunol.org/content/173/9/5343.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision http://www.jimmunol.org/ • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: by guest on October 1, 2021 http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2004 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. THE JOURNAL OF IMMUNOLOGY BRIEF REVIEWS Mechanisms of Soluble Cytokine Receptor Generation Stewart J. Levine1 Soluble cytokine receptors regulate inflammatory and im- biomarker of T cell activation and tumor burden in IL-2R␣- mune events by functioning as agonists or antagonists of expressing lymphoid neoplasms (3, 4). cytokine signaling. As such, they act within complex recep- Agonistic and antagonistic modulation of cytokine activity by soluble tor systems that include signaling receptors, nonsignaling cytokine receptors decoy receptors, receptor-associated proteins, and soluble The IL-1 and IL-6 receptor systems are paradigms for soluble receptor antagonists. -
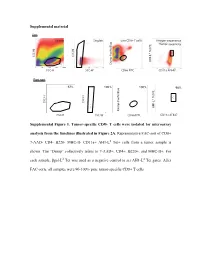
Supplemental Material Supplemental Figure 1. Tumor-Specific CD8+
Supplemental material Sort Scatter Singlets Live CD8+ T cells Antigen-experience Tumor-specifcity c Blue f Tet PE Tet d SSC-H SSC-H AH1-L Dump Paci FSC-H SSC-W CD8α FITC CD11a AF647 Post sort 67% 100%67% 100% 98% c Blue f Tet PE Tet d SSC-H SSC-H AH1-L SSC-Height Dump Paci FSC-H SSC-W CD8α FITC CD11a AF647 SSC-Height Supplemental Figure 1. Tumor-specific CD8+ T cells were isolated for microarray analysis from the timelines illustrated in Figure 2A. Representative FAC-sort of CD8+ 7-AAD- CD4- B220- MHC-II- CD11a+ AH1-Ld Tet+ cells from a tumor sample is shown. The “Dump” collectively refers to 7-AAD+, CD4+, B220+, and MHC-II+. For each sample, βgal-Ld Tet was used as a negative control to set AH1-Ld Tet gates. After FAC-sorts, all samples were 90-100% pure tumor-specific CD8+ T cells. Supplemental Table 1. Transcriptional profiles of published T cell programs compared to tumor-specific TIL T cell program of interest Exhausted (overexpressed) Exhausted (underrexpressed) Memory (overexpressed) Memory (underexpressed) Effector (overexpressed) Effector (underexpressed) Naïve virus-specific CD8+ T Naïve virus-specific CD8+ T Naïve virus-specific CD8+ Exhausted virus-specific CD8+ cells; also not in memory and Memory virus-specific CD8+ cells; also not in exhausted Effector virus-specific T cells; als not in memory, T cell programs compared for T cells vs. effector vs naïve virus-specific T cells vs. and effector vs naïve virus- CD8+ T cells vs. and exhausted vs naïve relative gene expression CD8+ T cells specific CD8+ T cells virus-specific CD8+ T cells Naïve virus-specific CD8+ T Naïve virus-specific CD8+ T Naïve virus-specific CD8+ T Naïve virus-specific CD8+ T Naïve virus-specific CD8+ Naïve virus-specific CD8+ cells cells cells cells T cells T cells Reference Wherry EJ et al.