Draft Genome Assembly and Annotation of the Gila Topminnow Poeciliopsis Occidentalis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
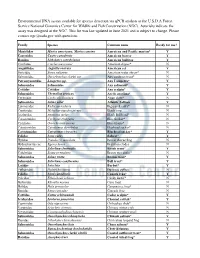
Edna Assay Development
Environmental DNA assays available for species detection via qPCR analysis at the U.S.D.A Forest Service National Genomics Center for Wildlife and Fish Conservation (NGC). Asterisks indicate the assay was designed at the NGC. This list was last updated in June 2021 and is subject to change. Please contact [email protected] with questions. Family Species Common name Ready for use? Mustelidae Martes americana, Martes caurina American and Pacific marten* Y Castoridae Castor canadensis American beaver Y Ranidae Lithobates catesbeianus American bullfrog Y Cinclidae Cinclus mexicanus American dipper* N Anguillidae Anguilla rostrata American eel Y Soricidae Sorex palustris American water shrew* N Salmonidae Oncorhynchus clarkii ssp Any cutthroat trout* N Petromyzontidae Lampetra spp. Any Lampetra* Y Salmonidae Salmonidae Any salmonid* Y Cottidae Cottidae Any sculpin* Y Salmonidae Thymallus arcticus Arctic grayling* Y Cyrenidae Corbicula fluminea Asian clam* N Salmonidae Salmo salar Atlantic Salmon Y Lymnaeidae Radix auricularia Big-eared radix* N Cyprinidae Mylopharyngodon piceus Black carp N Ictaluridae Ameiurus melas Black Bullhead* N Catostomidae Cycleptus elongatus Blue Sucker* N Cichlidae Oreochromis aureus Blue tilapia* N Catostomidae Catostomus discobolus Bluehead sucker* N Catostomidae Catostomus virescens Bluehead sucker* Y Felidae Lynx rufus Bobcat* Y Hylidae Pseudocris maculata Boreal chorus frog N Hydrocharitaceae Egeria densa Brazilian elodea N Salmonidae Salvelinus fontinalis Brook trout* Y Colubridae Boiga irregularis Brown tree snake* -

The Evolution of the Placenta Drives a Shift in Sexual Selection in Livebearing Fish
LETTER doi:10.1038/nature13451 The evolution of the placenta drives a shift in sexual selection in livebearing fish B. J. A. Pollux1,2, R. W. Meredith1,3, M. S. Springer1, T. Garland1 & D. N. Reznick1 The evolution of the placenta from a non-placental ancestor causes a species produce large, ‘costly’ (that is, fully provisioned) eggs5,6, gaining shift of maternal investment from pre- to post-fertilization, creating most reproductive benefits by carefully selecting suitable mates based a venue for parent–offspring conflicts during pregnancy1–4. Theory on phenotype or behaviour2. These females, however, run the risk of mat- predicts that the rise of these conflicts should drive a shift from a ing with genetically inferior (for example, closely related or dishonestly reliance on pre-copulatory female mate choice to polyandry in conjunc- signalling) males, because genetically incompatible males are generally tion with post-zygotic mechanisms of sexual selection2. This hypoth- not discernable at the phenotypic level10. Placental females may reduce esis has not yet been empirically tested. Here we apply comparative these risks by producing tiny, inexpensive eggs and creating large mixed- methods to test a key prediction of this hypothesis, which is that the paternity litters by mating with multiple males. They may then rely on evolution of placentation is associated with reduced pre-copulatory the expression of the paternal genomes to induce differential patterns of female mate choice. We exploit a unique quality of the livebearing fish post-zygotic maternal investment among the embryos and, in extreme family Poeciliidae: placentas have repeatedly evolved or been lost, cases, divert resources from genetically defective (incompatible) to viable creating diversity among closely related lineages in the presence or embryos1–4,6,11. -

Environmental Properties of Chemicals Volume 2
1 t ENVIRONMENTAL 1 PROTECTION Esa Nikunen . Riitta Leinonen Birgit Kemiläinen • Arto Kultamaa Environmental properties of chemicals Volume 2 1 O O O O O O O O OO O OOOOOO Ol OIOOO FINNISH ENVIRONMENT INSTITUTE • EDITA Esa Nikunen e Riitta Leinonen Birgit Kemiläinen • Arto Kultamaa Environmental properties of chemicals Volume 2 HELSINKI 1000 OlO 00000001 00000000000000000 Th/s is a second revfsed version of Environmental Properties of Chemica/s, published by VAPK-Pub/ishing and Ministry of Environment, Environmental Protection Department as Research Report 91, 1990. The pubiication is also available as a CD ROM version: EnviChem 2.0, a PC database runniny under Windows operating systems. ISBN 951-7-2967-2 (publisher) ISBN 952-7 1-0670-0 (co-publisher) ISSN 1238-8602 Layout: Pikseri Julkaisupalvelut Cover illustration: Jussi Hirvi Edita Ltd. Helsinki 2000 Environmental properties of chemicals Volume 2 _____ _____________________________________________________ Contents . VOLUME ONE 1 Contents of the report 2 Environmental properties of chemicals 3 Abbreviations and explanations 7 3.1 Ways of exposure 7 3.2 Exposed species 7 3.3 Fffects________________________________ 7 3.4 Length of exposure 7 3.5 Odour thresholds 8 3.6 Toxicity endpoints 9 3.7 Other abbreviations 9 4 Listofexposedspecies 10 4.1 Mammais 10 4.2 Plants 13 4.3 Birds 14 4.4 Insects 17 4.5 Fishes 1$ 4.6 Mollusca 22 4.7 Crustaceans 23 4.8 Algae 24 4.9 Others 25 5 References 27 Index 1 List of chemicals in alphabetical order - 169 Index II List of chemicals in CAS-number order -

Uncorrected Proofs for Review Only C5478.Indb 28 1/24/11 2:08:33 PM M
Chapter 3 Variation and evolution of reproductive strategies Marcelo N. Pires, Amanda Banet, Bart J. A. Pollux, and David N. Reznick 3.1 Introduction sociation between these two traits suggests that one of the two traits might be more likely to evolve when the other he family poeciliidae (Rosen & Bailey 1963) trait is already present (the latter facilitating the evolu- consists of a well-defi ned, monophyletic group of tion of the former). However, the existence of a notable Tnearly 220 species with a fascinating heterogene- exception in the literature (the lecithotrophic, superfetat- ity in life-history traits. Reznick and Miles (1989a) made ing Poeciliopsis monacha, the only known exception at the one of the fi rst systematic attempts to gather information time) showed that superfetation and matrotrophy were not from a widely scattered literature on poeciliid life histo- strictly linked, indicating that these two traits can evolve ries. They focused on two important female reproductive independently of each other. traits: (1) the ability to carry multiple broods at different Reznick and Miles (1989a) also proposed a framework developmental stages (superfetation; Turner 1937, 1940b, for future research that was aimed at evaluating possible 1940c), which tends to cause females to produce fewer off- causes and mechanisms for the evolution of superfetation spring per brood and to produce broods more frequently, and matrotrophy by (1) gathering detailed life-history de- and (2) the provisioning of eggs and developing embryos scriptions of a greater number of poeciliid species, either by the mother, which may occur prior to (lecithotrophy) or through common garden studies or from fi eld-collected after (matrotrophy) fertilization. -

Endangered Species
FEATURE: ENDANGERED SPECIES Conservation Status of Imperiled North American Freshwater and Diadromous Fishes ABSTRACT: This is the third compilation of imperiled (i.e., endangered, threatened, vulnerable) plus extinct freshwater and diadromous fishes of North America prepared by the American Fisheries Society’s Endangered Species Committee. Since the last revision in 1989, imperilment of inland fishes has increased substantially. This list includes 700 extant taxa representing 133 genera and 36 families, a 92% increase over the 364 listed in 1989. The increase reflects the addition of distinct populations, previously non-imperiled fishes, and recently described or discovered taxa. Approximately 39% of described fish species of the continent are imperiled. There are 230 vulnerable, 190 threatened, and 280 endangered extant taxa, and 61 taxa presumed extinct or extirpated from nature. Of those that were imperiled in 1989, most (89%) are the same or worse in conservation status; only 6% have improved in status, and 5% were delisted for various reasons. Habitat degradation and nonindigenous species are the main threats to at-risk fishes, many of which are restricted to small ranges. Documenting the diversity and status of rare fishes is a critical step in identifying and implementing appropriate actions necessary for their protection and management. Howard L. Jelks, Frank McCormick, Stephen J. Walsh, Joseph S. Nelson, Noel M. Burkhead, Steven P. Platania, Salvador Contreras-Balderas, Brady A. Porter, Edmundo Díaz-Pardo, Claude B. Renaud, Dean A. Hendrickson, Juan Jacobo Schmitter-Soto, John Lyons, Eric B. Taylor, and Nicholas E. Mandrak, Melvin L. Warren, Jr. Jelks, Walsh, and Burkhead are research McCormick is a biologist with the biologists with the U.S. -

Gila Topminnow Revised Recovery Plan December 1998
GILA TOPMINNOW, Poeciliopsis occidentalis occidentalis, REVISED RECOVERY PLAN (Original Approval: March 15, 1984) Prepared by David A. Weedman Arizona Game and Fish Department Phoenix, Arizona for Region 2 U.S. Fish and Wildlife Service Albuquerque, New Mexico December 1998 Approved: Regional Director, U.S. Fish and Wildlife Service Date: Gila Topminnow Revised Recovery Plan December 1998 DISCLAIMER Recovery plans delineate reasonable actions required to recover and protect the species. The U.S. Fish and Wildlife Service (Service) prepares the plans, sometimes with the assistance of recovery teams, contractors, State and Federal Agencies, and others. Objectives are attained and any necessary funds made available subject to budgetary and other constraints affecting the parties involved, as well as the need to address other priorities. Time and costs provided for individual tasks are estimates only, and not to be taken as actual or budgeted expenditures. Recovery plans do not necessarily represent the views nor official positions or approval of any persons or agencies involved in the plan formulation, other than the Service. They represent the official position of the Service only after they have been signed by the Regional Director or Director as approved. Approved recovery plans are subject to modification as dictated by new findings, changes in species status, and the completion of recovery tasks. ii Gila Topminnow Revised Recovery Plan December 1998 ACKNOWLEDGMENTS Original preparation of the revised Gila topminnow Recovery Plan (1994) was done by Francisco J. Abarca 1, Brian E. Bagley, Dean A. Hendrickson 1 and Jeffrey R. Simms 1. That document was modified to this current version and the work conducted by those individuals is greatly appreciated and now acknowledged. -

Understanding the Coexistence of Sperm-Dependent Asexual Species
Understanding the coexistence of sperm-dependent asexual species and their sexual hosts: the role of biogeography, mate choice, and relative fitness in the Phoxinus eos-neogaeus (Pisces: Cyprinidae) system by Jonathan Alan Mee B.Sc.F., The University of British Columbia, 2002 M.Sc., The University of Toronto, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Zoology) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) December 2011 © Jonathan Alan Mee, 2011 !"#$%&'$( In sperm-dependent asexual reproduction, sperm is not required for its genetic contribution, but it is required for stimulating zygote development. In my dissertation, I address several questions related to the coexistence of sperm-dependent asexuals and the sexually-reproducing species on which they depend. I have focused my research on a sperm-dependent asexual fish, Phoxinus eos-neogaeus, that originated via hybridization between P. eos and P. neogaeus. Using a mathematical model of mate choice among sexuals and sperm-dependent asexuals, I showed that stable coexistence can occur when there is variation among males in the strength of preference for mating with sexual females and when males with stronger preference pay a higher cost of preference. My model also predicts that coexistence is facilitated when the asexuals suffer a fitness disadvantage relative to the sexuals. Subsequent empirical work, in which I compared the repeat swimming performance, fecundity, and growth rate of asexual and sexual Phoxinus, provided results that are consistent with this prediction: the asexuals are, at best, as fit as the sexuals. I sampled Phoxinus populations from across the species’ North American distribution and the pattern of mitochondrial DNA variation across these populations suggests that all P. -

Food Habits of Imperiled Plains Topminnow and Diet Overlap With
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Transactions of the Nebraska Academy of Sciences Nebraska Academy of Sciences and Affiliated Societies Winter 3-1-2018 Food habits of imperiled Plains Topminnow and diet overlap with invasive Western Mosquitofish in the Central Great Plains Joseph Thiessen University of Nebraska at Kearney, [email protected] Keith D. Koupal Nebraska Game and Parks Commission, [email protected] Casey W. Schoenebeck University of Nebraska at Kearney, [email protected] Julie J. Shaffer University of Nebraska, Kearney Follow this and additional works at: https://digitalcommons.unl.edu/tnas Part of the Population Biology Commons, and the Terrestrial and Aquatic Ecology Commons Thiessen, Joseph; Koupal, Keith D.; Schoenebeck, Casey W.; and Shaffer, Julie J., "Food habits of imperiled Plains Topminnow and diet overlap with invasive Western Mosquitofish in the Central Great Plains" (2018). Transactions of the Nebraska Academy of Sciences and Affiliated Societies. 513. https://digitalcommons.unl.edu/tnas/513 This Article is brought to you for free and open access by the Nebraska Academy of Sciences at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Transactions of the Nebraska Academy of Sciences and Affiliated Societies by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Food habits of imperiled Plains Topminnow and diet overlap with invasive Western Mosquitofish in the Central Great Plains Joseph D. Thiessen,1,4 Keith D. Koupal,2 Casey W. Schoenebeck,1,3 and Julie J. Shaffer 1 1 Department of Biology, University of Nebraska at Kearney, Kearney, NE 68849 2 Nebraska Game and Parks Commission, Kearney Field Office, Kearney, NE 68847 3 Present address: Minnesota Department of Natural Resources, 23070 North Lakeshore Drive, Glenwood, MN 56334 4 Present address: Idaho Department of Fish and Game, 1414 E. -

Resistance and Resilience of Murray-Darling Basin Fishes to Drought Disturbance
Resistance and Resilience of Murray- Darling Basin Fishes to Drought Disturbance Dale McNeil1, Susan Gehrig1 and Clayton Sharpe2 SARDI Publication No. F2009/000406-1 SARDI Research Report Series No. 602 SARDI Aquatic Sciences PO Box 120 Henley Beach SA 5022 April 2013 Final Report to the Murray-Darling Basin Authority - Native Fish Strategy Project MD/1086 “Ecosystem Resilience and the Role of Refugia for Native Fish Communities & Populations” McNeil et. al. 2013 Drought and Native Fish Resilience Resistance and Resilience of Murray- Darling Basin Fishes to Drought Disturbance Final Report to the Murray-Darling Basin Authority - Native Fish Strategy Project MD/1086 “Ecosystem Resilience and the Role of Refugia for Native Fish Communities & Populations” Dale McNeil1, Susan Gehrig1 and Clayton Sharpe2 SARDI Publication No. F2009/000406-1 SARDI Research Report Series No. 602 April 2013 Page | ii McNeil et. al. 2013 Drought and Native Fish Resilience This Publication may be cited as: McNeil, D. G., Gehrig, S. L. and Sharpe, C. P. (2013). Resistance and Resilience of Murray-Darling Basin Fishes to Drought Disturbance. Final Report to the Murray-Darling Basin Authority - Native Fish Strategy Project MD/1086 ―Ecosystem Resilience and the Role of Refugia for Native Fish Communities & Populations‖. South Australian Research and Development Institute (Aquatic Sciences), Adelaide. SARDI Publication No. F2009/000406-1. SARDI Research Report Series No. 602. 143pp. Front Cover Images – Lake Brewster in the Lower Lachlan River catchment, Murray-Darling Basin during extended period of zero inflows, 2007. Murray cod (Maccullochella peelii peelii), olive perchlet (Ambassis agassizii) and golden perch (Macquaria ambigua) from the, lower Lachlan River near Lake Brewster, 2007 (all images - Dale McNeil). -

Poeciliidae: Poeciliopsis)
Copyright 0 1991 by the Genetics Society of America Molecular Evidence for Multiple Origins of Hybridogenetic Fish Clones (Poeciliidae: Poeciliopsis) Joseph M. Quattro,* John C. Aviset and Robert C. Vrijenhoek* *Center for Theoretical and Applied Genetics, Cook College, Rutgers University, New Brunswick, New Jersey 08903-0231, and ‘Department $Genetics, University of Georgia, Athens, Georgia 30602 Manuscript received September 6, 1990 Accepted for publication October 1 1, 1990 ABSTRACT Hybrid matings between the sexual species Poeciliopsis monacha and Poeciliopsis lucida produced a series of diploid all-female lineages of P. monacha-lucida that inhabit the Rio Fuerte of northwestern Mexico. Restriction site analyses of mitochondrial DNA (mtDNA) clearly revealed that P. monacha was the maternal ancestor of these hybrids. The high level of mtDNA diversity in P. monacha was mirrored by similarly high levels in P. monacha-lucida; thus hybridizations giving rise to unisexual lineages have occurred many times. However, mtDNA variability among P. monacha-lucida lineages revealed a geographical component. Apparently the opportunity for the establishment of unisexual lineages varies amongtributaries of the Rio Fuerte. We hypothesize thata dynamic complex of sexual and clonal fishes appear to participate in a feedback process that maintains genetic diversity in both the sexual and asexual components. ENETIC studies have revealed that clonally re- versity among the hemiclonal monacha genomes of P. G producing, all-female “species” of vertebrates monacha-lucida fromthe Rio Fuerte (VRIJENHOEK, are the products of hybridization between congeneric ANGUSand SCHULTZ1977). For brevity, we refer to sexual species (SCHULTZ 1969;SITES et al. 1990; VRI- distinct hemiclones defined by electrophoresis as “E- JENHOEK et al. -

Gila Chub (Gila Intermedia) DRAFT RECOVERY PLAN
Gila Chub (Gila intermedia) DRAFT RECOVERY PLAN Illustration credit: Randy Babb Southwest Region U.S. Fish and Wildlife Service Albuquerque, New Mexico Gila Chub (Gila intermedia) DRAFT RECOVERY PLAN Prepared by: Gila Chub Recovery Team, Technical Subgroup Prepared for: Southwest Region U.S. Fish and Wildlife Service Albuquerque, New Mexico DISCLAIMER Recovery plans delineate such reasonable actions as may be necessary, based upon the best scientific and commercial data available, for the conservation and survival of listed species. Plans are published by the U.S. Fish and Wildlife Service, and are sometimes prepared with the assistance of recovery teams, contractors, state agencies, and others. Recovery plans do not necessarily represent the views, official positions or approval of any individuals or agencies involved in the plan formulation, other than the U.S. Fish and Wildlife Service. They represent the official position of FWS only after they have been signed by the Regional Director. Recovery plans are guidance and planning documents only; identification of an action to be implemented by any public or private party does not create a legal obligation beyond existing legal requirements. Nothing in this plan should be construed as a commitment or requirement that any federal agency obligate or pay funds in any one fiscal year in excess of appropriations made by Congress for that fiscal year in contravention of the Anti-Deficiency Act, 31 U.S.C. 1341, or any other law or regulation. Approved recovery plans are subject to modification as dictated by new findings, changes in species status, and the completion of recovery actions. Literature citation of this document should read as follows: U.S. -

Status of Climate and Water Resources at Tumacácori National Historical Park Water Year 2016
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science Status of Climate and Water Resources at Tumacácori National Historical Park Water Year 2016 Natural Resource Report NPS/SODN/NRR—2017/1552 ON THE COVER Discharge sampling along the Santa Cruz River, Tumacácori National Historical Park. NPS photo. Status of Climate and Water Resources at Tumacácori National Historical Park Water Year 2016 Natural Resource Report NPS/SODN/NRR—2017/1552 Prepared by Evan L. Gwilliam Laura Palacios Sonoran Desert Network National Park Service 12661 E. Broadway Blvd. Tucson, AZ 85748 Kara Raymond Southern Arizona Office 3636 N. Central Ave., Suite 410 Phoenix, AZ 85012 Editing and Design Alice Wondrak Biel Sonoran Desert Network National Park Service 12661 E. Broadway Blvd. Tucson, AZ 85748 November 2017 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and oth- ers in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. The series supports the advancement of science, informed decision- making, and the achievement of the National Park Service mission. The series also provides a forum for presenting more lengthy results that may not be accepted by publications with page limitations.