Intramolecular Nicholas Reaction Enables the Stereoselective Synthesis of Strained Cyclooctynes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Rearrangements in the Camphor Series : the Decomposition Products of the Methyl Ester of Isoaminocamphonanic Acid
MOLECULAR REARRANGEMENTS IN THE CAMPHOR SERIES. THE DECOMPOSITION PRODUCTS OF THE METHYL ESTER OF ISOAMINOCAMPHONANIC ACID. BY GLENN SEYMOUR SKINNER A. B. Kansas State Manual Training Normal, 1913 A. M. University of Illinois, 1915 THESIS Submitted in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY IN CHEMISTRY IN THE GRADUATE SCHOOL OF THE UNIVERSITY OF ILLINOIS 1917 Digitized by the Internet Archive in 2013 http://archive.org/details/molecularrearranOOskin SK3 UNIVERSITY OF ILLINOIS THE GRADUATE SCHOOL .191 7 I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPER- VISION BY .QleiirL...S.eymo.nr.....S.]s;i.nner ENTITLED .MQlficulax....E.earrii.ng_meiits in... the .G.amj2hgr....Se^^^^ The I)ejG.QiQ.p.Q.ai.t.i.o.rL...I!r.Q.d.iL.c.t.s Q±....tha.JvIe.l]i3fl....B.st.e..r o.t.....Is..oanDn.ocam^^^^^ Acid. BE ACCEPTED AS FULFILLING THIS PART OF THE REQUIREMENTS FOR THE DEGREE OF.. .J).Q.C..1i.QX._..Q±....PJlilQ.S..Q.p.llZ. In ChargeCI of Thesis Head of Department Recommendation concurred in :* .<:. TSr:^. Committee on Final Examination* *Required for doctor's degree but not for master's. •^L^n .X o o TABLE OF GONTEIITS. PART I. HISTORICAL. The Camphors ------------------------- 1 The Camphoric Acids --------------------- 2 The Methyl Esters of d-Camphoric and 1-Isocaraphoric Acid - - - 3 The Methyl Esters of d-Camphoramidic and l-Isocamphoramidic Acids 4 The d-Oamphoramidic and 1- Is o camphoramidic Acids ------- 6 The Amino Acids Which are Derived from the d-Gamphoramidic and 1-Isocamphoramidic Acids ---------- 7 The Methyl Esters of the Amino Acid-S - -- -- -- -- -- -- 8 The Decomposition Products of Amino Camphonanic Acid ----- 8 The Decomposition Products of Dihydroaminocampholytic Acid - - 10 The Decomposition of Isoaminocamphonanic Acid ----- - 15 The Decomposition of Isodihydr oaminocampholytic Acid ----- 15 The Unsaturated Acids- -------------------- 1^ The Hydroxy Acids- ---------------------- 1^ The Hydrocarbons ----------------------- 22 The Lactones ------------------------- PART II. -

Gasket Chemical Services Guide
Gasket Chemical Services Guide Revision: GSG-100 6490 Rev.(AA) • The information contained herein is general in nature and recommendations are valid only for Victaulic compounds. • Gasket compatibility is dependent upon a number of factors. Suitability for a particular application must be determined by a competent individual familiar with system-specific conditions. • Victaulic offers no warranties, expressed or implied, of a product in any application. Contact your Victaulic sales representative to ensure the best gasket is selected for a particular service. Failure to follow these instructions could cause system failure, resulting in serious personal injury and property damage. Rating Code Key 1 Most Applications 2 Limited Applications 3 Restricted Applications (Nitrile) (EPDM) Grade E (Silicone) GRADE L GRADE T GRADE A GRADE V GRADE O GRADE M (Neoprene) GRADE M2 --- Insufficient Data (White Nitrile) GRADE CHP-2 (Epichlorohydrin) (Fluoroelastomer) (Fluoroelastomer) (Halogenated Butyl) (Hydrogenated Nitrile) Chemical GRADE ST / H Abietic Acid --- --- --- --- --- --- --- --- --- --- Acetaldehyde 2 3 3 3 3 --- --- 2 --- 3 Acetamide 1 1 1 1 2 --- --- 2 --- 3 Acetanilide 1 3 3 3 1 --- --- 2 --- 3 Acetic Acid, 30% 1 2 2 2 1 --- 2 1 2 3 Acetic Acid, 5% 1 2 2 2 1 --- 2 1 1 3 Acetic Acid, Glacial 1 3 3 3 3 --- 3 2 3 3 Acetic Acid, Hot, High Pressure 3 3 3 3 3 --- 3 3 3 3 Acetic Anhydride 2 3 3 3 2 --- 3 3 --- 3 Acetoacetic Acid 1 3 3 3 1 --- --- 2 --- 3 Acetone 1 3 3 3 3 --- 3 3 3 3 Acetone Cyanohydrin 1 3 3 3 1 --- --- 2 --- 3 Acetonitrile 1 3 3 3 1 --- --- --- --- 3 Acetophenetidine 3 2 2 2 3 --- --- --- --- 1 Acetophenone 1 3 3 3 3 --- 3 3 --- 3 Acetotoluidide 3 2 2 2 3 --- --- --- --- 1 Acetyl Acetone 1 3 3 3 3 --- 3 3 --- 3 The data and recommendations presented are based upon the best information available resulting from a combination of Victaulic's field experience, laboratory testing and recommendations supplied by prime producers of basic copolymer materials. -

Reactivity of Zinc Halide Complexes Containing Camphor-Derived Guanidine Ligands with Technical Rac-Lactide
inorganics Article Reactivity of Zinc Halide Complexes Containing Camphor-Derived Guanidine Ligands with Technical rac-Lactide Angela Metz 1, Joshua Heck 1, Clara Marie Gohlke 1, Konstantin Kröckert 1, Yannik Louven 1, Paul McKeown 2, Alexander Hoffmann 1, Matthew D. Jones 2 and Sonja Herres-Pawlis 1,* ID 1 Institute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany; [email protected] (A.M.); [email protected] (J.H.); [email protected] (C.M.G.); [email protected] (K.K.); [email protected] (Y.L.); [email protected] (A.H.) 2 Centre for Sustainable Chemical Technologies, Department of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK; [email protected] (P.M.); [email protected] (M.D.J.) * Correspondence: [email protected]; Tel.: +49-241-80-939-02 Received: 27 October 2017; Accepted: 23 November 2017; Published: 30 November 2017 Abstract: Three new zinc complexes with monoamine–guanidine hybridligands have been prepared, characterized by X-ray crystallography and NMR spectroscopy, and tested in the solvent-free ring-opening polymerization of rac-lactide. Initially the ligands were synthesized from camphoric acid to obtain TMGca and DMEGca and then reacted with zinc(II) halides to form zinc complexes. All complexes have a distorted tetrahedral coordination. They were utilized as catalysts in the solvent-free polymerization of technical rac-lactide at 150 ◦C. Colorless polylactide (PLA) can be produced and after 2 h conversion up to 60% was reached. -

EPDM & FKM Chemical Resistance Guide
EPDM & FKM Chemical Resistance Guide FIRST EDITION EPDM & FKM CHEMICAL RESISTANCE GUIDE Elastomers: Ethylene Propylene (EPDM) Fluorocarbon (FKM) Chemical Resistance Guide Ethylene Propylene (EPDM) & Fluorocarbon (FKM) 1st Edition © 2019 by IPEX. All rights reserved. No part of this book may be used or reproduced in any manner whatsoever without prior written permission. For information contact: IPEX, Marketing, 1425 North Service Road East, Oakville, Ontario, Canada, L6H 1A7 ABOUT IPEX At IPEX, we have been manufacturing non-metallic pipe and fittings since 1951. We formulate our own compounds and maintain strict quality control during production. Our products are made available for customers thanks to a network of regional stocking locations from coast-to-coast. We offer a wide variety of systems including complete lines of piping, fittings, valves and custom-fabricated items. More importantly, we are committed to meeting our customers’ needs. As a leader in the plastic piping industry, IPEX continually develops new products, modernizes manufacturing facilities and acquires innovative process technology. In addition, our staff take pride in their work, making available to customers their extensive thermoplastic knowledge and field experience. IPEX personnel are committed to improving the safety, reliability and performance of thermoplastic materials. We are involved in several standards committees and are members of and/or comply with the organizations listed on this page. For specific details about any IPEX product, contact our customer service department. INTRODUCTION Elastomers have outstanding resistance to a wide range of chemical reagents. Selecting the correct elastomer for an application will depend on the chemical resistance, temperature and mechanical properties needed. Resistance is a function both of temperatures and concentration, and there are many reagents which can be handled for limited temperature ranges and concentrations. -

List of Chemical Resistance of Materials in Contact with Liquid ANALYSETTE 22 Next & ANALYSETTE 28 Imagesizer LASER PARTICLE SIZER & PARTICLE SIZER
List of chemical resistance of materials in contact with liquid ANALYSETTE 22 NeXT & ANALYSETTE 28 ImageSizer LASER PARTICLE SIZER & PARTICLE SIZER This chart information is based on FRITSCH's knowledge and experience and are intended as a guide and not as a guarantee. We cannot assure the perfect performance of the materials as that depends on many factors as working pressure, pressure picks, fluid, ambient temperature, fluid temperature, concentra‐ tion, duration of exposure, etc… Fritsch GmbH Milling and Sizing Industriestrasse 8 D - 55743 Idar-Oberstein Telephone: +49 (0) 6784/ 70-0 Email: [email protected] Internet: www.fritsch.de Version 04/2020 Index 001 Table of contents Table of contents 1 Materials in contact with liquid.................................................... 4 2 Abbreviations................................................................................. 5 3 List of chemical resistance............................................................. 7 3.1 KEY......................................................................................... 7 3.2 Note....................................................................................... 7 3.3 Table 1.................................................................................... 7 3.4 Table 2.................................................................................. 97 3.5 Table 3................................................................................ 105 - 3 - Materials in contact with liquid 1 Materials in contact with liquid Wet dispersion -
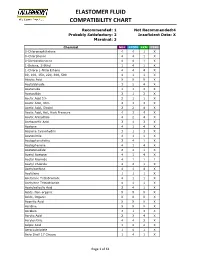
Elastomer Fluid Compatibility Chart
ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE 0-Chloronaphthalene 4 4 1 X 0-Chlorphenol 4 4 1 X 0-Dichlorobenzene 4 4 1 X 1-Butene, 2-Ethyl 1 4 1 X 1-Chloro 1-Nitro Ethane 4 4 4 X 90, 100, 150, 220, 300, 500 4 1 1 X Abietic Acid X X X X Acetaldehyde 3 2 4 X Acetamide 1 1 3 X Acetanilide 3 1 3 X Acetic Acid 5% 2 1 1 X Acetic Acid, 30% 2 1 3 X Acetic Acid, Glacial 2 2 4 X Acetic Acid, Hot, High Pressure 4 3 4 X Acetic Anhydride 4 2 4 X Acetoacetic Acid 3 1 3 X Acetone 4 1 4 X Acetone Cyanohydrin 3 1 3 X Acetonitrile 3 1 1 X Acetophenetidine 2 4 1 X Acetophenone 4 1 4 X Acetotoluidide 2 4 1 X Acetyl Acetone 4 1 4 X Acetyl Bromide 4 1 1 1 Acetyl Chloride 4 4 1 X Acetylacetone 4 1 4 X Acetylene 1 1 1 X Acetylene Tetrabromide 4 1 1 X Acetylene Tetrachloride 4 1 1 X Acetylsalicylic Acid 2 4 1 X Acids, Non-organic X X X X Acids, Organic X X X X Aconitic Acid X X X X Acridine X X X X Acrolein 3 1 3 X Acrylic Acid 2 3 4 X Acrylonitrile 4 4 3 X Adipic Acid 1 2 2 X Aero Lubriplate 1 4 1 X Aero Shell 17 Grease 1 4 1 X Page 1 of 61 ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE Aero Shell 1AC Grease 1 4 1 X Aero Shell 750 2 4 1 X Aero Shell 7A Grease 1 4 1 X Aerosafe 2300 4 1 4 X Aerosafe 2300W 4 1 4 X Aerozene 50 (50% Hydrazine 50% UDMH) 3 1 4 X Air, 1 1 1 X Air, 200 - 300° F 3 2 1 X Air, 300 - 400° F 4 4 1 X Air, 400 - 500° F 4 4 3 X Air, -

Organic Synthesis from France 10/02/2010
Baran Group Meeting Q. Michaudel Organic Synthesis from France 10/02/2010 Early French organic chemists (18th and 19th centuries): Albin Haller (1849–1925): Antoine-Laurent de Lavoisier (1743–1794): * semi-synthesis of camphor from camphoric acid and synthesis of menthol * law of conservation of mass * use of NaNH2 ("birth of strong bases chemistry") * recognized and named oxygen and hydrogen * abolished the phlogiston theory (see Seiple's GM 2010) Wurtz coupling (1855): * helped construct the metric system Na 2 + R W. heterocoupling * wrote the first extensive list of elements R1 X R2 X R1 * helped to reform chemical nomenclature THF, Et2O... Na R2 Wurtz-Fittig An appeal to delay the execution so that he could continue his experiments Ar X + R2 X Ar was cut short by the judge: "The Republic needs neither scientists nor THF, Et2O... chemists; the course of justice can not be delayed." (This quote may be more Ann. Chim. Phys. 1855, 96, 275 part of the legend than the reality) Lagrange's quote: "It took them only an instant to cut off his head, but France Müller modification: THF, -78°C, TPE (cat) to help Na solubilization may not produce another such head in a century." other modifications possible: different metal, sonication... Ph Ph Claude Berthollet (1748–1822): discovery of bleach Friedel-Crafts reaction (1877): Ph Ph Al, I2 Jean-Antoine Chaptal (1756–1832): TPE chaptalization (addition of sugar to unfermented wine to increase the Cl benzene I final alcohol level) Joseph Louis Gay-Lussac (1778–1850): " !!! * work in thermodynamics (Gay-Lussac's law) * discovery of boron, cyanogen and hydrogen cyanide Compt. -

(+)-Camphoric Acid Pierre Thuéry, Jack Harrowfield
Chiral one- to three-dimensional uranyl-organic assemblies from (1R,3S)-(+)-camphoric acid Pierre Thuéry, Jack Harrowfield To cite this version: Pierre Thuéry, Jack Harrowfield. Chiral one- to three-dimensional uranyl-organic assemblies from (1R,3S)-(+)-camphoric acid. CrystEngComm, Royal Society of Chemistry, 2014, 16 (14), pp.2996- 3004. 10.1039/c3ce42613k. hal-01157641 HAL Id: hal-01157641 https://hal.archives-ouvertes.fr/hal-01157641 Submitted on 17 Nov 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. CrystEngComm View Article Online PAPER View Journal | View Issue Chiral one- to three-dimensional uranyl–organic assemblies from (1R,3S)-(+)-camphoric acid† Cite this: CrystEngComm,2014,16, 2996 Pierre Thuéry*a and Jack Harrowfieldb Four complexes were obtained from reaction of uranyl nitrate with (1R,3S)-(+)-camphoric acid under solvo-/hydrothermal conditions with either acetonitrile or N-methyl-2-pyrrolidone (NMP) as the organic component. All complexes crystallize in chiral space groups and are enantiopure species. Complexes [(UO2)4(L)3(OH)2(H2O)4]·3H2O(1) and [(UO2)8K8(L)12(H2O)12]·H2O(2) were obtained in water–acetonitrile in the presence of LiOH or KOH in excess beyond or equal to that simply required to neutralize the acid, respectively. -

Supplement Of
Supplement of Feedbacks between acidity and multiphase chemistry of atmospheric aqueous particles and clouds 5 Andreas Tilgner1, Thomas Schaefer1, Becky Alexander2, Mary Barth3, Jeffrey L. Collett, Jr.4, Kathleen M. Fahey5, Athanasios Nenes6,7, Havala O. T. Pye5, Hartmut Herrmann1*, and V. Faye McNeill8* 1Leibniz Institute for Tropospheric Research (TROPOS), Atmospheric Chemistry Department (ACD), Leipzig, 04318, Germany 2Department of Atmospheric Science, University of Washington, Seattle, WA, 98195, USA 10 3National Center for Atmospheric Research, Boulder, CO, 80307, USA 4Department of Atmospheric Science, Colorado State University, Fort Collins, CO, 80523, USA 5Office of Research and Development, US Environmental Protection Agency, Research Triangle Park, NC, 27711, USA 6 School of Architecture, Civil and Environmental Engineering, Ecole Polytechnique Fédérale de Lausanne, Lausanne, CH- 1015, Switzerland 15 7Institute for Chemical Engineering Sciences, Foundation for Research and Technology Hellas, Patras, GR-26504, Greece 8Department of Chemical Engineering, Columbia University, New York, NY, 10027, USA Correspondence to: V. Faye McNeill ([email protected]), H. Herrmann ([email protected]) 1 Supplementary material to section 2 20 Table S1: Summary of the applied !"($) and &!' values for calculation of the LWC-acidity dependent aqueous fraction ($')presented in Figure 2, Figure 3 and Figure S1. KH Compound Reference pKa1 pKa2 Reference (298K) M atm-1 Sulfurous acid 1.32 Sander (2015) 1.9 7.0 Kolthoff and Elving (1959) (dissolved SO2) Nitrous acid 48.6 Park and Lee (1988) 3.29 — Kolthoff and Elving (1959) HONO Formic acid 5530 Khan et al. (1992) 3.77 — Braude et al. (1955) HCOOH Acetic acid 5471 Khan et al. (1992) 4.76 — Haynes (1958) CH3COOH Glycolic acid 2.83·104 Ip et al. -

William Albert Noyes
NATIONAL ACADEMY OF SCIENCES WILLIAM ALBERT NOYES 1857—1941 A Biographical Memoir by ROGER ADAMS Any opinions expressed in this memoir are those of the author(s) and do not necessarily reflect the views of the National Academy of Sciences. Biographical Memoir COPYRIGHT 1952 NATIONAL ACADEMY OF SCIENCES WASHINGTON D.C. WILLIAM ALBERT NOYES1 BY ROGER ADAMS William Albert Noyes was born November 6, 1857 on a farm near Independence, Iowa. He was the seventh generation of the Noyes family in America, being descended from Nicholas Noyes who came with his elder brother, James, from Choulder- ton, Wiltshire, in 1633. Their father had been a clergyman but had died before the sons left England. The two brothers settled first in Medford, Massachusetts but in 1655 moved to Newbury. Samuel, of the third generation in this direct line, moved to Abington about 1713 and there he and the succeeding generations lived for over one hundred and forty years. Spencer W. Noyes, the great grandson of Samuel, was ad- mitted as a student to Phillips Academy at Andover, Massachu- setts, March 21, 1837. He married Mary Packard-related to the famous family of shoe manufacturers-and they had three children when they left: Abington and migrated to Iowa in 185j. When the father with his family went west to open up a home- stead, he found conditions typical of the prairies and life was necessarily primitive. In the east he had been a cobbler by trade so had had little experience working the land. The early years were difficult ones because of the hard work in establishing a farm home, getting land into cultivation, raising food and storing sufficient for winter needs. -

Derivatives of Isocamphoric Acid, Decomposition of Isodihydroaminocampholytic Acid with Nitrous Acid
THE UNIVERSITY OF ILLINOIS LIBRARY DERIVATIVES OF ISOCAMPHORIC ACID, DECOMPOSI- TION OF ISODIHYDROAMINOCAMPHOLYTIC ACID WITH NITROUS ACID BY LLOYD FRANCIS NICKELL A. B. University of Illinois, 1909. A. M. University of Illinois, 1911. THESIS Submitted in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY IN CHEMISTRY IN THE GRADUATE SCHOOL OF THE UNIVERSITY OF ILLINOIS 1913 UNIVERSITY OF ILLINOIS THE GRADUATE SCHOOL May 17, i9$3, I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPERVISION BY Lloyd Franois Niakell ENTITLED Derivatives of Iaooamphoris Asid, Decomposition of Isodihydro- a~ino3anpholyti3 Aoid with Nitrous Aoid, BE ACCEPTED AS FULFILLING THIS PART OF THE REQUIREMENTS FOR THE DEGREE OF Dootor of Philosophy. In Charge of Maj or Work Head of DepartmentI Recommendation concurred in: Committee on Final Examination 247428 Digitized by the Internet Archive in 2013 http://archive.org/details/derivativesofisoOOnick DERIVATIVES OF ISOCAMPHORIG ACID DECOMPOSITION OF ISODIHYDROAIvIINOCAMPHOLYTIC ACID WITH NITROUS ACID UlUC This investigation was carried on during the academic year 1912 - 1913. It was undertaken at the suggestion of Profe33or William A . Noyes, ani. carried cut under his direction. The writer wishes to express his deep appreciation not only for the helpful suggestions which Professor Noyes has always teen so ready and so willing to give, but more especially for the point of view and the lasting inspiration for a life work which only close contact with a sincere, sympa- thetic teacher can give. In addition the writer wishes to ac- knowledge the kindness of Mr. R. S. Potter and of Mr. P. S. Woodward in giving him samples of desired materials. -

Analysis of Camphor and Camphoric Acid
Philosophical Magazine Series 2 ISSN: 1941-5850 (Print) 1941-5869 (Online) Journal homepage: http://www.tandfonline.com/loi/tphm13 Analysis of camphor and camphoric acid M. Liebig To cite this article: M. Liebig (1832) Analysis of camphor and camphoric acid, Philosophical Magazine Series 2, 11:62, 149-150, DOI: 10.1080/14786443208647703 To link to this article: http://dx.doi.org/10.1080/14786443208647703 Published online: 25 Jun 2009. Submit your article to this journal Article views: 2 View related articles Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=tphm13 Download by: [RMIT University Library] Date: 05 April 2016, At: 02:22 Intelligence and Miscellaneous Articles. 149 lain furnace. More lately Gmelin attributed this reduetion loerse to the presence of gaseous oxide of carbon in the furnace, and assigned the same cause for the reduction of oxide of iron in a porcelain furnace observed by Proust. In tact, it appears con- tradictory that a metal which oxidizes so readily as nickel, by cal- cination in the air, which burns in oxygen gas with disengagement of light, and is even susceptible of spontaneous combustion at corn- man temperatures when it is much divided, should be reduced from its oxide merely by the action of a strong heat. Nevertheless, it does not appear that direct experiments have been made on this subject, although it has often been remarked that during the re. duction of nickel in porcelain furnaces, without the use of charcoal, less metal was always obtained when the crucible was well closed.