Fluorine Containing Phosphine Complexes of The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Vibrationally Excited Hydrogen Halides : a Bibliography On
VI NBS SPECIAL PUBLICATION 392 J U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards National Bureau of Standards Bldg. Library, _ E-01 Admin. OCT 1 1981 191023 / oO Vibrationally Excited Hydrogen Halides: A Bibliography on Chemical Kinetics of Chemiexcitation and Energy Transfer Processes (1958 through 1973) QC 100 • 1X57 no. 2te c l !14 c '- — | NATIONAL BUREAU OF STANDARDS The National Bureau of Standards' was established by an act of Congress March 3, 1901. The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau consists of the Institute for Basic Standards, the Institute for Materials Research, the Institute for Applied Technology, the Institute for Computer Sciences and Technology, and the Office for Information Programs. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consistent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry, and commerce. The Institute consists of a Center for Radiation Research, an Office of Meas- urement Services and the following divisions: Applied Mathematics — Electricity — Mechanics — Heat — Optical Physics — Nuclear Sciences" — Applied Radiation 2 — Quantum Electronics 1 — Electromagnetics 3 — Time 3 1 1 and Frequency — Laboratory Astrophysics — Cryogenics . -

Phosphine and Ammonia Photochemistry in Jupiter's
40th Lunar and Planetary Science Conference (2009) 1201.pdf PHOSPHINE AND AMMONIA PHOTOCHEMISTRY IN JUPITER’S TROPOSPHERE. C. Visscher1, A. D. Sperier2, J. I. Moses1, and T.C. Keane3. 1Lunar and Planetary Institute, USRA, 3600 Bay Area Blvd., Houston, TX 77058-1113 2Baylor University, Waco, TX 76798, 3Department of Chemistry and Physics, The Sage Colleges, Troy, NY 12180, ([email protected], [email protected]) Introduction: The last comprehensive photo- tions involving the amino radical (NH2) play a larger chemical model for Jupiter’s troposphere was pre- role. We note that this is the first photochemical sented by Edgington et al. [1,2], based upon the work model to include P2H as an intermediate species in PH3 of Atreya et al. [3] and Kaye and Strobel [4-6]. Since photolysis. The equivalent N2Hx species are believed these early studies, numerous laboratory experiments to be important in the combustion chemistry of nitro- have led to an improvement in our understanding of gen compounds [15]. PH3, NH3-PH3, and NH3-C2H2 photochemistry [7-13]. Furthermore, recent Galileo, Cassini, and Earth-based observations have better defined the abundance of key atmospheric constituents as a function of altitude and latitude in Jupiter’s troposphere. These new results provide an opportunity to test and improve theoretical models of Jovian atmospheric chemistry. We have therefore developed a photochemical model for Jupi- ter’s troposphere considering the updated experimental and observational constraints. Using the Caltech/JPL KINETICS code [14] for our photochemical models, our basic approach is two- fold. The validity of our selected chemical reaction list is first tested by simulating the laboratory experiments of PH3, NH3-PH3, and NH3-C2H2 photolysis with pho- tochemical “box” models. -

UNITED STATES PATENT of FICE 2,640,086 PROCESS for SEPARATING HYDROGEN FLUORIDE from CHLORODFLUORO METHANE Robert H
Patented May 26, 1953 2,640,086 UNITED STATES PATENT of FICE 2,640,086 PROCESS FOR SEPARATING HYDROGEN FLUORIDE FROM CHLORODFLUORO METHANE Robert H. Baldwin, Chadds Ford, Pa., assignor to E. H. du Pont de Nemours and Company, Wi inington, Del, a corporation of Delaware No Drawing. Application December 15, 1951, Serial No. 261,929 9 Claims. (C. 260-653) 2 This invention relates to a process for Sep These objects are accomplished essentially by arating hydrogen fluoride from monochlorodi Subjecting a mixture of hydrogen fluoride and fluoronethane, and more particularly, separat Inonochlorodifluoromethane in the liquid phase ing these components from the reaction mixture to temperatures below 0° C., preferably at about obtained in the fluorination of chloroform with -30° C. to -50° C., at either atmospheric or hydrogen fluoride, Super-atmospheric pressures, together with from In the fluorination of chloroform in the prest about 0.25 mol to about 2.5 mols of chloroform ence Of a Catalyst, a reaction mixture is pro per mol of chlorodifluoronethane contained in duced which consists essentially of HCl, HF, the mixture and separating an upper layer rich CHCIF2, CHCl2F, CHCls, and CHF3. A method O in HF from a lower organic layer. The proceSS of Separating these components is disclosed in is operative with mixtures containing up to 77% U. S. Patent No. 2,450,414 which involves sep by weight of HF. arating the components by a special fractional It has been found that chloroform is substan distillation under appropriate temperatures and tially immiscible With EIF at temperatures be pressures. -
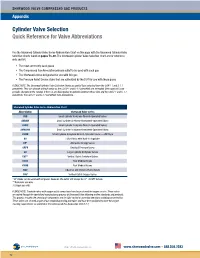
Cylinder Valve Selection Quick Reference for Valve Abbreviations
SHERWOOD VALVE COMPRESSED GAS PRODUCTS Appendix Cylinder Valve Selection Quick Reference for Valve Abbreviations Use the Sherwood Cylinder Valve Series Abbreviation Chart on this page with the Sherwood Cylinder Valve Selection Charts found on pages 73–80. The Sherwood Cylinder Valve Selection Chart are for reference only and list: • The most commonly used gases • The Compressed Gas Association primary outlet to be used with each gas • The Sherwood valves designated for use with this gas • The Pressure Relief Device styles that are authorized by the DOT for use with these gases PLEASE NOTE: The Sherwood Cylinder Valve Selection Charts are partial lists extracted from the CGA V-1 and S-1.1 pamphlets. They can change without notice as the CGA V-1 and S-1.1 pamphlets are amended. Sherwood will issue periodic changes to the catalog. If there is any discrepancy or question between these lists and the CGA V-1 and S-1.1 pamphlets, the CGA V-1 and S-1.1 pamphlets take precedence. Sherwood Cylinder Valve Series Abbreviation Chart Abbreviation Sherwood Valve Series AVB Small Cylinder Acetylene Wrench-Operated Valves AVBHW Small Cylinder Acetylene Handwheel-Operated Valves AVMC Small Cylinder Acetylene Wrench-Operated Valves AVMCHW Small Cylinder Acetylene Handwheel-Operated Valves AVWB Small Cylinder Acetylene Wrench-Operated Valves — WB Style BV Hi/Lo Valves with Built-in Regulator DF* Alternative Energy Valves GRPV Residual Pressure Valves GV Large Cylinder Acetylene Valves GVT** Vertical Outlet Acetylene Valves KVAB Post Medical Valves KVMB Post Medical Valves NGV Industrial and Chrome-Plated Valves YVB† Vertical Outlet Oxygen Valves 1 * DF Valves can be used with all gases; however, the outlet will always be ⁄4"–18 NPT female. -

Ammonium Bifluoride CAS No
Product Safety Summary Ammonium Bifluoride CAS No. 1341-49-7 This Product Safety Summary is intended to provide a general overview of the chemical substance. The information on the summary is basic information and is not intended to provide emergency response information, medical information or treatment information. The summary should not be used to provide in-depth safety and health information. In-depth safety and health information can be found in the Safety Data Sheet (SDS) for the chemical substance. Names • Ammonium bifluoride (ABF) • Ammonium difluoride • Ammonium acid fluoride • Ammonium hydrogen difluoride • Ammonium fluoride compound with hydrogen fluoride (1:1) Product Overview Solvay Fluorides, LLC does not sell ammonium bifluoride directly to consumers. Ammonium bifluoride is used in industrial applications and in other processes where workplace exposures can occur. Ammonium bifluoride (ABF) is used for cleaning and etching of metals before they are further processed. It is used as an oil well acidifier and in the etching of glass or cleaning of brick and ceramics. It may also be used for pH adjustment in industrial textile processing or laundries. ABF is available as a solid or liquid solution (in water). Ammonium bifluoride is a corrosive chemical and contact can severely irritate and burn the skin and eyes causing possible permanent eye damage. Breathing ammonium bifluoride can severely irritate and burn the nose, throat, and lungs, causing nosebleeds, cough, wheezing and shortness of breath. On contact with water or moist skin, ABF can release hydrofluoric acid, a very dangerous acid. Inhalation or ingestion of large amounts of ammonium bifluoride can cause nausea, vomiting and loss of appetite. -

Hexafluorosilicic Acid
Sodium Hexafluorosilicate [CASRN 16893-85-9] and Fluorosilicic Acid [CASRN 16961-83-4] Review of Toxicological Literature October 2001 Sodium Hexafluorosilicate [CASRN 16893-85-9] and Fluorosilicic Acid [CASRN 16961-83-4] Review of Toxicological Literature Prepared for Scott Masten, Ph.D. National Institute of Environmental Health Sciences P.O. Box 12233 Research Triangle Park, North Carolina 27709 Contract No. N01-ES-65402 Submitted by Karen E. Haneke, M.S. (Principal Investigator) Bonnie L. Carson, M.S. (Co-Principal Investigator) Integrated Laboratory Systems P.O. Box 13501 Research Triangle Park, North Carolina 27709 October 2001 Toxicological Summary for Sodium Hexafluorosilicate [16893-85-9] and Fluorosilicic Acid [16961-83-4] 10/01 Executive Summary Nomination Sodium hexafluorosilicate and fluorosilicic acid were nominated for toxicological testing based on their widespread use in water fluoridation and concerns that if they are not completely dissociated to silica and fluoride in water that persons drinking fluoridated water may be exposed to compounds that have not been thoroughly tested for toxicity. Nontoxicological Data Analysis and Physical-Chemical Properties Analytical methods for sodium hexafluorosilicate include the lead chlorofluoride method (for total fluorine) and an ion-specific electrode procedure. The percentage of fluorosilicic acid content for water supply service application can be determined by the specific-gravity method and the hydrogen titration method. The American Water Works Association (AWWA) has specified that fluorosilicic acid contain 20 to 30% active ingredient, a maximum of 1% hydrofluoric acid, a maximum of 200 mg/kg heavy metals (as lead), and no amounts of soluble mineral or organic substance capable of causing health effects. -

Occupational Exposure to Hydrogen Fluoride
criteria for a recommended standard OCCUPATIONAL EXPOSURE TO HYDROGEN FLUORIDE U.S. DEPARTMENT OF HEALTH, EDUCATION, AND WELFARE Public Health Service Center for Disease Control National Institute for Occupational Safety and Health M arch 1976 HEW Publication No. (NIOSH) 7 6 -1 4 3 PREFACE The Occupational Safety and Health Act of 1970 emphasizes the need for standards to protect the health and safety of workers exposed to an ever-increasing number of potential hazards at their workplace. The National Institute for Occupational Safety and Health has projected a formal system of research, with priorities determined on the basis of specified indices, to provide relevant data from which valid criteria for effective standards can be derived. Recommended standards for occupational exposure, which are the result of this work, are based on the health effects of exposure. The Secretary of Labor will weigh these recommen dations along with other considerations such as feasibility and means of implementation in developing regulatory standards. It is intended to present successive reports as research and epide miologic studies are completed and as sampling and analytical methods are developed. Criteria and standards will be reviewed periodically to ensure continuing protection of the worker. I am pleased to acknowledge the contributions to this report on hydrogen fluoride by members of my staff and the valuable constructive comments by the Review Consultants on Hydrogen Fluoride, by the ad hoc committees of the American Academy of Occupational Medicine and the Society for Occupational and Environmental Health, and by Robert B. O'Connor, M.D., NIOSH consultant in occupational medicine. -

Hydrogen Fluoride-Catalyzed Addition of Carbon Monoxide to Propylene Under High Pressures*
Hydrogen Fluoride-Catalyzed Addition of Carbon Monoxide to Propylene under High Pressures* Yoshimasa Takezaki**, Yoshio Fuchigami**, Hiroshi Teranishi**, Nobuyuki Sugita** and Kiyoshi Kudo** Summary: Isobutyric acid forming reaction of propylene with carbon monoxide has been studied in the presence of HF-H2O catalyst under high pressures. The optimum conditions for the formation of acid have been decided as follows: water content of HF catalyst, 20 wt. %; charge ratio of HF to C3H6, 15 (mole ratio);reaction temperature,94℃ or lower;and the total pressure,190kg/cm2 or higher. Reaction temperature higher than 94℃ is unfavorable because of accelerated polymerization of C3H6. Reaction mechanism of the formation of isobutyric acid has been proposed, where the rate determining step, C3H7++CO(dissolved)→C3H7CO+, being first-order with regard to each reactant. The rate expression for the yield of the acid has been derived and the apparent activation energy of the over-all reaction has been found to be 21.7kcal/mole. Introduction Since 1933 when several patents1) on the production of carboxylic acids from olefins and carbon monoxide in acidic media were In these works Koch obtained good yield published, many works have been carried (better than 90%) of acids when butene or out2)-22)on this process. On propylene as an higher olefin was used8),9), but in the case of olefin, the first systematic study was reported propylene5), detailed data on experimental by Hardy in 19362), in which the author, conditions and yields were not reported except using 87 %-phosphoric acid as the catalyst, for the formations of alcohols, esters and observed 50% yield of total acids (isobutyric carboxylic acids as the products. -

United States Patent Office
Patented June 26, 195i 2558,703 UNITED STATES PATENT OFFICE PREPARATION OFPROPENE TRIFLUOROTRICHLORO Carl I. Gochenour, Niagara Falls, N. Y., assignor to Hooker Electrochemical Company, Niagara Falls, N. Y., a corporation of New York No Drawing. Application January 6, 1948, Serial No. 818 5 Claims. (CI. 260-653) 2 pene are unsatisfactory; when more than six The present invention relates to a method for moles of hydrogen fluoride to one mole of hexa the fluorination of hexachloropropene, and is chloropropene are employed, yields of trifluoro more particularly concerned with the fluorination trichloropropene are decreased by the entrain of hexachloropropene to produce high yields of ment of product in the escaping excess HF. Also, ... trifluorotrichloropropene, and especially 1,1,1-tri the use of more than 6 moles is economically fluoro-2,3,3-trichloro-2,3-propene impractical. (CF3-CCECC) 3. The presence of pentavalent antimony It has been proposed to fluorinate hexachloro halide in catalytic amounts, from about 0.2 to 2.0 propene with antimony (III) fluoride and a cata 10 per cent by weight of the hexachloropropene be lyst, and this method of fluorination, although ing preferred. Representative catalysts are anti operative, is too expensive for economic .com mony (V), chloride, antimony (V) bromide, anti mercialization. It has also been proposed previ mony (V) fluoride, and antimony (V) chlorofluo-. ously to fluorinate hexachloropropene by heating rides. The antimony (V) chloride or chloroflu it together with a fluoromethane and an alumi 5 orides are preferred. Less than about 0.2 per num halide to accomplish exchange of the flu cent of catalyst is usually not as satisfactory for orine atom or atoms in the fluoromethane and practical yields; more than about 2.0 per cent halogen atoms other than fluorine in the hexa does not appreciably increase the yield. -

Phosphine Safety Data Sheet (180KB)
Phosphine Safety Data Sheet P-4643 This SDS conforms to U.S. Code of Federal Regulations 29 CFR 1910.1200, Hazard Communication. Date of issue: 01/01/1980 Revision date: 08/31/2018 Supersedes: 10/24/2016 SECTION: 1. Product and company identification 1.1. Product identifier Product form : Substance Substance name : Phosphine CAS-No. : 7803-51-2 Formula : PH3 Other means of identification : Hydrogen phosphide, Phosphorous hydride, Phosphorous trihydride, Phosphorated hydrogen 1.2. Relevant identified uses of the substance or mixture and uses advised against Use of the substance/mixture : Industrial use; Use as directed. 1.3. Details of the supplier of the safety data sheet Praxair, Inc. 10 Riverview Drive Danbury, CT 06810-6268 - USA T 1-800-772-9247 (1-800-PRAXAIR) - F 1-716-879-2146 www.praxair.com 1.4. Emergency telephone number Emergency number : Onsite Emergency: 1-800-645-4633 CHEMTREC, 24hr/day 7days/week — Within USA: 1-800-424-9300, Outside USA: 001-703-527-3887 (collect calls accepted, Contract 17729) SECTION 2: Hazard identification 2.1. Classification of the substance or mixture GHS-US classification Pyr. Gas H250 Flam. Gas 1 H220 Press. Gas (Liq.) H280 Acute Tox. 1 (Inhalation:gas) H330 Skin Corr. 1B H314 Eye Dam. 1 H318 Aquatic Acute 1 H400 2.2. Label elements GHS-US labeling Hazard pictograms (GHS-US) : GHS02 GHS04 GHS05 GHS06 GHS09 Signal word (GHS-US) : Danger Hazard statements (GHS-US) : H220 - EXTREMELY FLAMMABLE GAS H250 - CATCHES FIRE SPONTANEOUSLY IF EXPOSED TO AIR H280 - CONTAINS GAS UNDER PRESSURE; MAY EXPLODE IF HEATED H314 - Causes severe skin burns and eye damage H330 - FATAL IF INHALED H400 - VERY TOXIC TO AQUATIC LIFE CGA-HG04 - MAY FORM EXPLOSIVE MIXTURES WITH AIR CGA-HG11 - SYMPTOMS MAY BE DELAYED Precautionary statements (GHS-US) : P202 - Do not handle until all safety precautions have been read and understood. -

NATURE [APRIL 9, 1932 Having for Comparison a Standard Benzene Bulb
546 NATURE [APRIL 9, 1932 having for comparison a standard benzene bulb. filter transmitting 250-280 p.p. was used in these ex The powder was then melted, and recrystallised by periments. The effect was shown to have its origin heating the bulb in an electric heater to about 270° C. in the phosphine molecule, but in view of the fact and very slowly cooling it. After noting the defl.exion that phosphine itself does not absorb in the region due to the bulb with the resolidified bismuth, that of 250-280 p.p., it would seem that the incidental presence the container alone was determined by breaking it of mercury vapour resulted in the phosphine being open and dissolving out the metal with nitric acid. decomposed by excited mercury atoms into hydrogen Experiments with similar bulbs and bismuth regulus atoms and probably PH or PH2 radicals, which pro showed no change of defl.exion after heat treatment. duce the Hinshelwood-Clusius effect. Hinshelwood But when bismuth powder was melted and recrystal and Clusius concluded that the active material was lised, the diamagnetic susceptibility rose nearly to the present in the gas phase, but on repeating their ex regulus value. Many samples of colloidal bismuth periment by exposing the mixture in one tube and gave similar results. determining the explosion pressure immediately after These observations indicate that the fall in the in another similar tube, the effect was not observed. susceptibility value with reduction of particle size in That is, the effect is most probably a wall phenomenon, the case of bismuth is a genuine effect. -

DECOMPOSITION PRODUCTS of FLUOROCARBON POLYMERS
criteria for a recommended standard . occupational exposure to DECOMPOSITION PRODUCTS of FLUOROCARBON POLYMERS U.S. DEPARTMENT OF HEALTH, EDUCATION, AND WELFARE criteria for a recommended standard... OCCUPATIONAL EXPOSURE TO DECOMPOSITION PRODUCTS of FLUOROCARBON POLYMERS U.S. DEPARTMENT OF HEALTH, EDUCATION, AND WELFARE Public Health Service Center for Disease Control National Institute for Occupational Safety and Health September 1977 For sale by the Superintendent of Documents, U.S. Government Printing Office, Washington, D.C. 20402 DHEW (NIOSH) Publication No. 77-193 PREFACE The Occupational Safety and Health Act of 1970 emphasizes the need for standards to protect the health and safety of workers exposed to an ever-increasing number of potential hazards at their workplace. The National Institute for Occupational Safety and Health has projected a formal system of research, with priorities determined on the basis of specified indices, to provide relevant data from which valid criteria for effective standards can be derived. Recommended standards for occupational exposure, which are the result of this work, are based on the health effects of exposure. The Secretary of Labor will weigh these recommendations along with other considerations such as feasibility and means of implementation in developing regulatory standards. It is intended to present successive reports as research and epidemiologic studies are completed and as sampling and analytical methods are developed. Criteria and standards will be reviewed periodically to ensure continuing protection of the worker. I am pleased to acknowledge the contributions to this report on the decomposition products of fluorocarbon polymers by members of the NIOSH staff and the valuable constructive comments by the Review Consultants on the Decomposition Products of Fluorocarbon Polymers and by Robert B.