Genetic Network Identifies Novel Pathways Contributing To
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Information
doi: 10.1038/nature08795 SUPPLEMENTARY INFORMATION Supplementary Discussion Population naming In some contexts, the indigenous hunter-gatherer and pastoralist peoples of southern Africa are referred to collectively as the Khoisan (Khoi-San) or more recently Khoesan (Khoe-San) people. This grouping is based on the unique linguistic use of click-consonants1. Many names, often country-specific, have been used by Bantu pastoralists and European settlers to describe the hunter-gatherers, including San, Saan, Sonqua, Soaqua, Souqua, Sanqua, Kwankhala, Basarwa, Batwa, Abathwa, Baroa, Bushmen, Bossiesmans, Bosjemans, or Bosquimanos. In addition, group-specific names such as !Kung and Khwe are often used for the broader population. The two most commonly used names, “San” and “Bushmen”, have both been associated with much controversy due to derogatory connotations2. “San” has become the more popular term used in Western literature, although “Bushmen” is arguably the more commonly recognized term within the communities. Since they have no collective name for themselves, the term Bushmen was selected for use in this paper as the term most familiar to the participants themselves. Regarding identification of individuals The five men identified in this study have all elected to have their identity made public knowledge. Thus we present two complete personal genomes (KB1 and ABT), a low-coverage personal genome (NB1), and personal exomes for all five men. On a scientific level, identification allows for current and future correlation of genetic data with demographic and medical histories. On a social level, identification allows for maximizing community benefit. For !Gubi, G/aq’o, D#kgao and !Aî, their name represents not only themselves, but importantly their extended family unit and a way of life severely under threat. -

Analyses of Allele-Specific Gene Expression in Highly Divergent
ARTICLES Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance James J Crowley1,10, Vasyl Zhabotynsky1,10, Wei Sun1,2,10, Shunping Huang3, Isa Kemal Pakatci3, Yunjung Kim1, Jeremy R Wang3, Andrew P Morgan1,4,5, John D Calaway1,4,5, David L Aylor1,9, Zaining Yun1, Timothy A Bell1,4,5, Ryan J Buus1,4,5, Mark E Calaway1,4,5, John P Didion1,4,5, Terry J Gooch1,4,5, Stephanie D Hansen1,4,5, Nashiya N Robinson1,4,5, Ginger D Shaw1,4,5, Jason S Spence1, Corey R Quackenbush1, Cordelia J Barrick1, Randal J Nonneman1, Kyungsu Kim2, James Xenakis2, Yuying Xie1, William Valdar1,4, Alan B Lenarcic1, Wei Wang3,9, Catherine E Welsh3, Chen-Ping Fu3, Zhaojun Zhang3, James Holt3, Zhishan Guo3, David W Threadgill6, Lisa M Tarantino7, Darla R Miller1,4,5, Fei Zou2,11, Leonard McMillan3,11, Patrick F Sullivan1,5,7,8,11 & Fernando Pardo-Manuel de Villena1,4,5,11 Complex human traits are influenced by variation in regulatory DNA through mechanisms that are not fully understood. Because regulatory elements are conserved between humans and mice, a thorough annotation of cis regulatory variants in mice could aid in further characterizing these mechanisms. Here we provide a detailed portrait of mouse gene expression across multiple tissues in a three-way diallel. Greater than 80% of mouse genes have cis regulatory variation. Effects from these variants influence complex traits and usually extend to the human ortholog. Further, we estimate that at least one in every thousand SNPs creates a cis regulatory effect. -

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

Rabbit Anti-NEURL2 Antibody-SL19213R
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-NEURL2 antibody SL19213R Product Name: NEURL2 Chinese Name: E3Ubiquitin连接酶蛋白NEURL2抗体 Neuralized E3 Ubiquitin Protein Ligase 2; C20orf163; Chromosome 20 Open Reading Alias: Frame 163; Neuralized Homolog 2 (Drosophila) ; Neuralized-Like 2 (Drosophila); Neuralized-Like Protein 2; Neuralized Homolog 2; OZZ-E3; OZZ; NEUL2_HUMAN; Organism Species: Rabbit Clonality: Polyclonal React Species: Human,Mouse,Rat,Dog,Pig,Cow,Horse,Rabbit,Sheep, ELISA=1:500-1000IHC-P=1:400-800IHC-F=1:400-800ICC=1:100-500IF=1:100- 500(Paraffin sections need antigen repair) Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 32kDa Cellular localization: cytoplasmic Form: Lyophilized or Liquid Concentration: 1mg/ml immunogen: KLHwww.sunlongbiotech.com conjugated synthetic peptide derived from human NEURL2:1-100/285 Lsotype: IgG Purification: affinity purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. PubMed: PubMed This gene encodes a protein that is involved in the regulation of myofibril organization. This protein is likely the adaptor component of the E3 ubiquitin ligase complex in Product Detail: striated muscle, and it regulates the ubiquitin-mediated degradation of beta-catenin during myogenesis. -

Datasheet Blank Template
SAN TA C RUZ BI OTEC HNOL OG Y, INC . Neuralized-2 (J-24): sc-133814 BACKGROUND APPLICATIONS Neuralized-2, also known as NEURL2, OZZ or Ozz-E3, is a 285 amino acid Neuralized-2 (J-24) is recommended for detection of Neuralized-2 of human cytoplasmic protein involved in myofiber differentiation and maturation that origin by Western Blotting (starting dilution 1:200, dilution range 1:100- is induced during differentiation. Expressed predominantly in skeletal and 1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein (1 ml cardiac muscle, Neuralized-2 localizes to the tips of differentiating myofibers of cell lysate)] and solid phase ELISA (starting dilution 1:30, dilution range and regulates ubiquitination and degradation of β-catenin at the sarcolem ma. 1:30-1:3000). Neuralized-2 contains one SOCS box domain, through which it interacts with Suitable for use as control antibody for Neuralized-2 siRNA (h): sc-75905, Elongin B and Elongin C, and one NHR (neuralized homology repeat) domain Neuralized-2 shRNA Plasmid (h): sc-75905-SH and Neuralized-2 shRNA (h) that is implicated in ubiquitination substrate binding. The gene encoding Lentiviral Particles: sc-75905-V. Neuralized-2 maps to human chromosome 20, which is comprised of approx i- mately 2% of the human genome, contains nearly 63 million bases and Molecular Weight of Neuralized-2: 31 kDa. encodes over 600 genes. Creutzfeldt-Jakob disease, amyotrophic lateral sclerosis and Alagille syndrome are associated with chromosome 20. RECOMMENDED SECONDARY REAGENTS To ensure optimal results, the following support (secondary) reagents are REFERENCES recommended: 1) Western blotting: use goat anti-rabbit IgG-HRP: sc-2004 1. -

Systematic Elucidation of Neuron-Astrocyte Interaction in Models of Amyotrophic Lateral Sclerosis Using Multi-Modal Integrated Bioinformatics Workflow
ARTICLE https://doi.org/10.1038/s41467-020-19177-y OPEN Systematic elucidation of neuron-astrocyte interaction in models of amyotrophic lateral sclerosis using multi-modal integrated bioinformatics workflow Vartika Mishra et al.# 1234567890():,; Cell-to-cell communications are critical determinants of pathophysiological phenotypes, but methodologies for their systematic elucidation are lacking. Herein, we propose an approach for the Systematic Elucidation and Assessment of Regulatory Cell-to-cell Interaction Net- works (SEARCHIN) to identify ligand-mediated interactions between distinct cellular com- partments. To test this approach, we selected a model of amyotrophic lateral sclerosis (ALS), in which astrocytes expressing mutant superoxide dismutase-1 (mutSOD1) kill wild-type motor neurons (MNs) by an unknown mechanism. Our integrative analysis that combines proteomics and regulatory network analysis infers the interaction between astrocyte-released amyloid precursor protein (APP) and death receptor-6 (DR6) on MNs as the top predicted ligand-receptor pair. The inferred deleterious role of APP and DR6 is confirmed in vitro in models of ALS. Moreover, the DR6 knockdown in MNs of transgenic mutSOD1 mice attenuates the ALS-like phenotype. Our results support the usefulness of integrative, systems biology approach to gain insights into complex neurobiological disease processes as in ALS and posit that the proposed methodology is not restricted to this biological context and could be used in a variety of other non-cell-autonomous communication -

Comparative Analysis of the Ubiquitin-Proteasome System in Homo Sapiens and Saccharomyces Cerevisiae
Comparative Analysis of the Ubiquitin-proteasome system in Homo sapiens and Saccharomyces cerevisiae Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Universität zu Köln vorgelegt von Hartmut Scheel aus Rheinbach Köln, 2005 Berichterstatter: Prof. Dr. R. Jürgen Dohmen Prof. Dr. Thomas Langer Dr. Kay Hofmann Tag der mündlichen Prüfung: 18.07.2005 Zusammenfassung I Zusammenfassung Das Ubiquitin-Proteasom System (UPS) stellt den wichtigsten Abbauweg für intrazelluläre Proteine in eukaryotischen Zellen dar. Das abzubauende Protein wird zunächst über eine Enzym-Kaskade mit einer kovalent gebundenen Ubiquitinkette markiert. Anschließend wird das konjugierte Substrat vom Proteasom erkannt und proteolytisch gespalten. Ubiquitin besitzt eine Reihe von Homologen, die ebenfalls posttranslational an Proteine gekoppelt werden können, wie z.B. SUMO und NEDD8. Die hierbei verwendeten Aktivierungs- und Konjugations-Kaskaden sind vollständig analog zu der des Ubiquitin- Systems. Es ist charakteristisch für das UPS, daß sich die Vielzahl der daran beteiligten Proteine aus nur wenigen Proteinfamilien rekrutiert, die durch gemeinsame, funktionale Homologiedomänen gekennzeichnet sind. Einige dieser funktionalen Domänen sind auch in den Modifikations-Systemen der Ubiquitin-Homologen zu finden, jedoch verfügen diese Systeme zusätzlich über spezifische Domänentypen. Homologiedomänen lassen sich als mathematische Modelle in Form von Domänen- deskriptoren (Profile) beschreiben. Diese Deskriptoren können wiederum dazu verwendet werden, mit Hilfe geeigneter Verfahren eine gegebene Proteinsequenz auf das Vorliegen von entsprechenden Homologiedomänen zu untersuchen. Da die im UPS involvierten Homologie- domänen fast ausschließlich auf dieses System und seine Analoga beschränkt sind, können domänen-spezifische Profile zur Katalogisierung der UPS-relevanten Proteine einer Spezies verwendet werden. Auf dieser Basis können dann die entsprechenden UPS-Repertoires verschiedener Spezies miteinander verglichen werden. -

Developmental Biology Catalog
ptglab.com 1 ANTIBODIES FOR DEVELOPMENTAL BIOLOGY www.ptglab.com 2 Antibodies For Developmental Biology Front Cover: IF result (trunk or trunck-associated region; nucleus stain; RED) of NKX2-2 antibody (13013-1-AP) with E16.5 mouse pancreas by Dr. Nicholas George, Sarvetnick Lab – UNMC. (Green, E-Cadherin; RED, NKX2-2; Blue, DAPI). ptglab.com 3 WELCOME Foreword Developmental biology covers a broad spectrum of scientific research relating to the growth and development of living things. Not only does it concern the embryogenic events immediately following fertilization, it also encompasses the genetic control of cell growth, differentiation and morphogenesis – key components of regeneration and aging in the adult organism. In this development-focused catalog This catalog is essentially a shortlist you will find antibodies to those targets of around one quarter of the antibodies involved in pattern formation, such as Proteintech has for developmental biology HOX gene products and proteins involved protein targets. If you can’t find an antibody in Notch and Hedgehog signaling; neural to your target of choice here, we’re confident tube formation, such as sonic hedgehog that with over 2,000 primary antibodies protein; and organogenesis such as Wnt, relating to development in the complete FGF, BMP and EYA proteins. At the center Proteintech inventory you’ll find what you’re of the catalog, you will also find a primer looking for online at www.ptglab.com. on the primary cilium, with details on Proteintech®* antibodies recognizing the proteins involved in the generation and maintenance of this vital developmental structure. What’s Inside 6–7 Focus Article Antibodies For Cilia Development 8 Focus Article Investigating Kidney Development With Proteintech’s SIX2 Antibody 9–11 Antibodies: ABLIM1 GSK3B 12–13 Antibodies For Cilia Development 14–17 Antibodies: GSK3B ZHX2 18 Contact Us Please Note: All products featured in this catalog are for research use only. -

MAFB Determines Human Macrophage Anti-Inflammatory
MAFB Determines Human Macrophage Anti-Inflammatory Polarization: Relevance for the Pathogenic Mechanisms Operating in Multicentric Carpotarsal Osteolysis This information is current as of October 4, 2021. Víctor D. Cuevas, Laura Anta, Rafael Samaniego, Emmanuel Orta-Zavalza, Juan Vladimir de la Rosa, Geneviève Baujat, Ángeles Domínguez-Soto, Paloma Sánchez-Mateos, María M. Escribese, Antonio Castrillo, Valérie Cormier-Daire, Miguel A. Vega and Ángel L. Corbí Downloaded from J Immunol 2017; 198:2070-2081; Prepublished online 16 January 2017; doi: 10.4049/jimmunol.1601667 http://www.jimmunol.org/content/198/5/2070 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2017/01/15/jimmunol.160166 Material 7.DCSupplemental References This article cites 69 articles, 22 of which you can access for free at: http://www.jimmunol.org/content/198/5/2070.full#ref-list-1 by guest on October 4, 2021 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2017 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. -
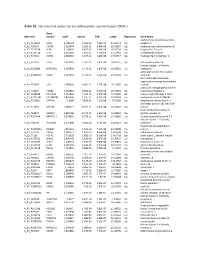
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

The Pdx1 Bound Swi/Snf Chromatin Remodeling Complex Regulates Pancreatic Progenitor Cell Proliferation and Mature Islet Β Cell
Page 1 of 125 Diabetes The Pdx1 bound Swi/Snf chromatin remodeling complex regulates pancreatic progenitor cell proliferation and mature islet β cell function Jason M. Spaeth1,2, Jin-Hua Liu1, Daniel Peters3, Min Guo1, Anna B. Osipovich1, Fardin Mohammadi3, Nilotpal Roy4, Anil Bhushan4, Mark A. Magnuson1, Matthias Hebrok4, Christopher V. E. Wright3, Roland Stein1,5 1 Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, TN 2 Present address: Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN 3 Department of Cell and Developmental Biology, Vanderbilt University, Nashville, TN 4 Diabetes Center, Department of Medicine, UCSF, San Francisco, California 5 Corresponding author: [email protected]; (615)322-7026 1 Diabetes Publish Ahead of Print, published online June 14, 2019 Diabetes Page 2 of 125 Abstract Transcription factors positively and/or negatively impact gene expression by recruiting coregulatory factors, which interact through protein-protein binding. Here we demonstrate that mouse pancreas size and islet β cell function are controlled by the ATP-dependent Swi/Snf chromatin remodeling coregulatory complex that physically associates with Pdx1, a diabetes- linked transcription factor essential to pancreatic morphogenesis and adult islet-cell function and maintenance. Early embryonic deletion of just the Swi/Snf Brg1 ATPase subunit reduced multipotent pancreatic progenitor cell proliferation and resulted in pancreas hypoplasia. In contrast, removal of both Swi/Snf ATPase subunits, Brg1 and Brm, was necessary to compromise adult islet β cell activity, which included whole animal glucose intolerance, hyperglycemia and impaired insulin secretion. Notably, lineage-tracing analysis revealed Swi/Snf-deficient β cells lost the ability to produce the mRNAs for insulin and other key metabolic genes without effecting the expression of many essential islet-enriched transcription factors.