September 11, 2018 DUR Minutes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DRUGS REQUIRING PRIOR AUTHORIZATION in the MEDICAL BENEFIT Page 1
Effective Date: 08/01/2021 DRUGS REQUIRING PRIOR AUTHORIZATION IN THE MEDICAL BENEFIT Page 1 Therapeutic Category Drug Class Trade Name Generic Name HCPCS Procedure Code HCPCS Procedure Code Description Anti-infectives Antiretrovirals, HIV CABENUVA cabotegravir-rilpivirine C9077 Injection, cabotegravir and rilpivirine, 2mg/3mg Antithrombotic Agents von Willebrand Factor-Directed Antibody CABLIVI caplacizumab-yhdp C9047 Injection, caplacizumab-yhdp, 1 mg Cardiology Antilipemic EVKEEZA evinacumab-dgnb C9079 Injection, evinacumab-dgnb, 5 mg Cardiology Hemostatic Agent BERINERT c1 esterase J0597 Injection, C1 esterase inhibitor (human), Berinert, 10 units Cardiology Hemostatic Agent CINRYZE c1 esterase J0598 Injection, C1 esterase inhibitor (human), Cinryze, 10 units Cardiology Hemostatic Agent FIRAZYR icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent HAEGARDA c1 esterase J0599 Injection, C1 esterase inhibitor (human), (Haegarda), 10 units Cardiology Hemostatic Agent ICATIBANT (generic) icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent KALBITOR ecallantide J1290 Injection, ecallantide, 1 mg Cardiology Hemostatic Agent RUCONEST c1 esterase J0596 Injection, C1 esterase inhibitor (recombinant), Ruconest, 10 units Injection, lanadelumab-flyo, 1 mg (code may be used for Medicare when drug administered under Cardiology Hemostatic Agent TAKHZYRO lanadelumab-flyo J0593 direct supervision of a physician, not for use when drug is self-administered) Cardiology Pulmonary Arterial Hypertension EPOPROSTENOL (generic) -

TEXAS MEDICAID Clinical Edit Prior Authorization Epoetin Alfa (PROCRIT)
TEXAS MEDICAID Clinical Edit Prior Authorization epoetin alfa (PROCRIT) STEP 1: CLEARLY PRINT AND COMPLETE TO EXPEDITE PROCESSING Date: Prescriber First & Last Name: Patient First & Last Name: Prescriber NPI: Patient Address: Prescriber Address: Patient ID: Prescriber Phone: Patient Date of Birth: Prescriber Fax: STEP 2: MEDICATION INFORMATION Medication Requested (Name): Quantity Requested: Dose Requested: Dosing Instructions: Patient’s Primary Diagnosis: ____________________________________ ICD 10 Code: __________ Please indicate ONE (1) of the following: OR STAR / STAR KIDS client (Go to Step 3 - PDL PA Criteria Applies) OR CHIP / PERINATE client (Go to Step 4) STEP 3: PDL PRIOR AUTHORIZATION CRITERIA FOR NON-PREFERRED PRODUCT 1. Has the client failed a 30-day treatment trial with at least 1 preferred agent in the last 180 days? Yes (Go to Step 4 Question 1) No (Go to #2) 2. Is there a documented allergy or contraindication to preferred agents in this class? Yes (Go to Step 4 Question 1) No (Go to #3) 3. Is the drug necessary for treatment of stage-4 advanced metastatic cancer and associated conditions? Yes (Go to Step 4 Question 1) No (Deny) Rev. 11/18/2020 Page 1 of 3 Version 1.5 STEP 4: CLINICAL PRIOR AUTHORIZATION CRITERIA 1. Does the client have a diagnosis of chronic renal failure in the last 730 days? Yes (Go to #7) No (Go to #2) 2. Does the client have a diagnosis of cancer in the last 730 days? Yes (Go to #3) No (Go to #5) 3. Does the client have a history of an antineoplastic agent in the last 30 days? Examples of antineoplastic -

UWHC Guidelines for the Use of Darbepoetin and Epoetin
Use of Darbepoetin and Epoetin in Non-Nephrology Patients – Adult/Pediatric – Inpatient/Ambulatory Clinical Practice Guideline Table of Contents Executive Summary ......................................................................................... 3 Scope ............................................................................................................. 7 Methodology ................................................................................................... 7 Definitions (optional): ...................................................................................... 8 Introduction.................................................................................................... 8 Recommendations........................................................................................... 9 UW Health Implementation............................................................................ 19 References ................................................................................................... 19 Note: Active Table of Contents Click to follow link CPG Contact for Changes: Name: Philip Trapskin, PharmD, BCPS, Manager, DPP Phone Number: 608-263-1328 Email address: [email protected] CPG Contact for Content: Name: Jason Bergsbaken, PharmD Phone Number: 608-265-0341 Email address: [email protected] Copyright © 2015 University of Wisconsin Hospitals and Clinics Authority Contact: [email protected] Vermeulen, [email protected] Last Revised: 07/2015 Guideline Authors: Jason Bergsbaken, PharmD Coordinating -

Dr. Winegarden Presentation
Empowering Market Competition through Biosimilars Presentation to National Council of Insurance Legislators (NCOIL) 2019 Summer Meeting Newport Beach, California July 12, 2019 Total Savings (in millions) 25% 50% 75% Originator Total Annual Savings for Current, 25%, Drug Class Current Biosimilar Biosimilar Biosimilar Biologic 50%, and 75% Biosimilar Share Share Share Share Scenarios Compared to All-Originator Infliximab Remicade $79.4 $318.2 $636.5 $954.7 Biologic Baseline Pegfilgrastim Neulasta $21.8 $121.9 $243.8 $365.7 Filgrastim Neupogen $152.1 $152.1 $152.1 $206.8 Epoetin Alfa Epogen & Procrit $0.5 $8.4 $16.9 $25.3 Bevacizumab Avastin $0.0 $199.2 $398.5 $597.7 Trastuzumab Herceptin $0.0 $208.0 $415.9 $623.9 • Biosimilars price discounts expected 30% - Rituxumab Rituxan $0.0 $280.6 $561.2 $841.8 40% off originator biologic Etanercept Enbrel $0.0 $324.0 $648.0 $972.1 • Forthcoming study estimates possible Adalimunab Humira $0.0 $861.1 $1,722.1 $2,583.2 health care savings from wider adoption of biosimilars. GRAND TOTAL $253.8 $2,473.6 $4,795.0 $7,171.2 • Estimates are based on the average sales price (ASP) data that are effective from April 2019 through June 2019 and rolling 12-month volume data through February 2019. Biosimilars have no “clinically meaningful difference” in safety, purity, • Methodology: compare potential savings to hypothetical all-originator biologic scenario and effectiveness relative to its reference originator biologic. Just like • Over 10 years, potential savings of $24.7 generic medicines, the benefits of biosimilars are the substantial price billion, $48.0 billion, and $71.7 billion respectively. -
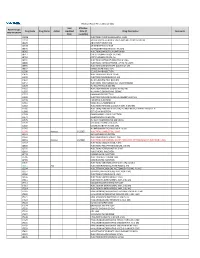
2021 Prior Authorization List Part B Appendix a (PDF)
Medicare Part B PA List Effective 2021 Last Effective Part B Drugs: Drug Code Drug Name Action Updated Date (if Drug Description Comments STEP THERAPY Date available) C9050 INJECTION, EMAPALUMAB-LZSG, 1 MG C9122 MOMETASONE FUROATE SINUS IMPLANT 10 MCG SINUVA J0129 ABATACEPT INJECTION J0178 AFLIBERCEPT INJECTION J0570 BUPRENORPHINE IMPLANT 74.2MG J0585 INJECTION,ONABOTULINUMTOXINA J0717 CERTOLIZUMAB PEGOL INJ 1MG J0718 CERTOLIZUMAB PEGOL INJ J0791 INJECTION CRIZANLIZUMAB-TMCA 5 MG J0800 INJECTION, CORTICOTROPIN, UP TO 40 UNITS J0896 INJECTION LUSPATERCEPT-AAMT 0.25 MG J0897 DENOSUMAB INJECTION J1300 ECULIZUMAB INJECTION J1428 INJECTION ETEPLIRSEN 10 MG J1429 INJECTION GOLODIRSEN 10 MG J1442 INJ FILGRASTIM EXCL BIOSIMIL J1447 INJECTION, TBO-FILGRASTIM, 1 MICROGRAM J1459 INJ IVIG PRIVIGEN 500 MG J1555 INJECTION IMMUNE GLOBULIN 100 MG J1556 INJ, IMM GLOB BIVIGAM, 500MG J1557 GAMMAPLEX INJECTION J1558 INJECTION IMMUNE GLOBULIN XEMBIFY 100 MG J1559 HIZENTRA INJECTION J1561 GAMUNEX-C/GAMMAKED J1562 INJECTION; IMMUNE GLOBULIN 10%, 5 GRAMS J1566 INJECTION, IMMUNE GLOBULIN, INTRAVENOUS, LYOPHILIZED (E.G. P J1568 OCTAGAM INJECTION J1569 GAMMAGARD LIQUID INJECTION J1572 FLEBOGAMMA INJECTION J1575 INJ IG/HYALURONIDASE 100 MG IG J1599 IVIG NON-LYOPHILIZED, NOS J1602 GOLIMUMAB FOR IV USE 1MG J1745 INJ INFLIXIMAB EXCL BIOSIMILR 10 MG J1930 Remove 1/1/2021 INJECTION, LANREOTIDE, 1 MG J2323 NATALIZUMAB INJECTION J2350 INJECTION OCRELIZUMAB 1 MG J2353 Remove 1/1/2021 INJECTION, OCTREOTIDE, DEPOT FORM FOR INTRAMUSCULAR INJECTION, 1 MG J2357 INJECTION, OMALIZUMAB, -

Presentation Title
The COMMANDS trial: a phase 3 study of the efficacy and safety of luspatercept versus epoetin alfa for the treatment of anemia due to Revised International Prognostic Scoring System Very Low-, Low-, or Intermediate-risk myelodysplastic syndromes in erythropoiesis stimulating agent-naive patients who require red blood cell transfusions Matteo Della Porta,1,2 Uwe Platzbecker,3 Valeria Santini,4 Guillermo Garcia-Manero,5 Rami S. Komrokji,6 Rodrigo Ito,7 Pierre Fenaux8 1Cancer Center IRCCS Humanitas Research Hospital, Milan, Italy; 2Department of Biomedical Sciences, Humanitas University, Milan, Italy; 3Medical Clinic and Policlinic 1, Hematology and Cellular Therapy, University Hospital Leipzig, Leipzig, Germany; 4Azienda Ospedaliero-Universitaria Careggi, University of Florence, Florence, Italy; 5Department of Leukemia, University of Texas MD Anderson Cancer Center, Houston, TX; 6Moffitt Cancer Center, Tampa, FL; 7Bristol Myers Squibb, Princeton, NJ; 8Service d'Hématologie Séniors, Hôpital Saint-Louis, Université Paris 7, Paris, France Presentation 2198 Presenting author disclosures M.D.P.: no conflicts of interest to disclose. 2 Introduction and objectives Introduction • Studies of epoetin alfa and darbepoetin alfa have demonstrated efficacy among patients with LR-MDS, but the patient population in which a clinically significant effect is observed may be limited1,2 • Luspatercept, a first-in-class erythroid maturation agent with a mechanism of action distinct from ESAs,3 is approved by the US FDA for the treatment of anemia failing an -

MEDICATION GUIDE PROCRIT® (PRO′−KRIT) (Epoetin Alfa)
MEDICATION GUIDE PROCRIT® (PRO′−KRIT) (epoetin alfa) Read this Medication Guide: • before you start PROCRIT. • if you are told by your healthcare provider that there is new information about PROCRIT. • if you are told by your healthcare provider that you may inject PROCRIT at home, read this Medication Guide each time you receive a new supply of medicine. This Medication Guide does not take the place of talking to your healthcare provider about your medical condition or your treatment. Talk with your healthcare provider regularly about the use of PROCRIT and ask if there is new information about PROCRIT. What is the most important information I should know about PROCRIT? Using PROCRIT can lead to death or other serious side effects. For patients with cancer: Your healthcare provider has received special training through the ESA APPRISE Oncology Program in order to prescribe PROCRIT. Before you can begin to receive PROCRIT, you must sign the patient-healthcare provider acknowledgment form. When you sign this form, you are stating that your healthcare provider talked with you about the risks of taking PROCRIT. These risks include that your tumor may grow faster and you may die sooner if you choose to take PROCRIT. You should talk with your healthcare provider about: • Why PROCRIT treatment is being prescribed for you. • What are the chances you will get red blood cell transfusions if you do not take PROCRIT. • What are the chances you will get red blood cell transfusions even if you take PROCRIT. • How taking PROCRIT may affect the success of your cancer treatment. -

Epoetin Alfa)
MEDICATION GUIDE PROCRIT® (PRO′−KRIT) (epoetin alfa) Read this Medication Guide before you start PROCRIT, each time you refill your prescription, and if you are told by your healthcare provider that there is new information about PROCRIT. This Medication Guide does not take the place of talking to your healthcare provider about your medical condition or your treatment. Talk with your healthcare provider regularly about the use of PROCRIT and ask if there is new information about PROCRIT . What is the most important information I should know about PROCRIT ? Using PROCRIT can lead to death or other serious side effects. Patients with cancer: Your healthcare provider has received special training through the ESA APPRISE Oncology Program in order to prescribe PROCRIT. Before you can begin to receive PROCRIT, you must sign the ESA APPRISE Oncology Patient and Healthcare Professional (HCP) Acknowledgement Form to document that your healthcare provider discussed the risks of PROCRIT with you. When you sign this form, you are stating that you are aware of the risks associated with use of PROCRIT. These risks include that your tumor may grow faster and you may die sooner when PROCRIT is used experimentally to try to raise your hemoglobin beyond the amount needed to avoid red blood cell transfusion or if you are not getting strong doses of chemotherapy. It is not known whether these risks exist when PROCRIT is given according to the FDA-approved directions for use. You should discuss with your doctor: • Why PROCRIT treatment is being prescribed. • What are the chances you will get red blood cell transfusions if you do not take PROCRIT . -

Historical Background
Historical Background In 1836, Richard Bright first commented on the pallor of patients with poor kidney function. In 1918, Profes- sor Paul Carnot (1869 – 1957), Paris, France, suggested a hormonal link to renal anemia and postulated a sub- stance “hemopoietin” which stimulated red cell produc- tion. Other scientists researched further [1, 2, 3]. In 1948, two Finnish scientists renamed the hormonal substance “erythropoietin” (EPO) [4]. In 1950, Reisman showed that the hypoxic rat caused erythropoieses in its parabi- otic, non-hypoxic partner [5]. In 1953, Erslev demon- strated a hormone in anemic animals which stimulated erythropoiesis when injected into non-anemic recipients [6]. Research between 1950 until 1970 revealed the kid- neys as the main production site [7]. In 1988, Koury and Lacombe detected significant EPO-levels in the peritu- bular interstitium by cDNA-analysis [8, 9]. The highest concentrations were found in the proximal convoluted tu- bule and in the cortical ascending loop of Henle. In 1989, epoetin alfa was first licensed and, in the same year, the EPO-receptor (EPO-R) was described [10]. 2 Anemia of Chronic Renal Failure Definition of Anemia The WHO defines anemia in general as hemoglobin < 13 g/dl in males and < 12 g/dl in females [11]. Similarly, KDOQI defines anemia as Hb < 13.5 g/dl in adult males and < 12.0 g/dl in adult females. The European Best Practice Guidelines for patients with renal disease has defined anemia as hemoglobin < 13.5 g/dl in males younger than 70 years, < 12.0 g/ dl in males > 70 years and < 11.5 g/dl in females of any age [12]. -

Novel Erythropoiesis-Stimulating Agents: a New Era in Anemia Management
Mini-Review Novel Erythropoiesis-Stimulating Agents: A New Era in Anemia Management Iain C. Macdougall Department of Renal Medicine, King’s College Hospital, London, United Kingdom Nearly two decades ago, recombinant human erythropoietin transformed the management of chronic kidney disease anemia by allowing a more sustained increase in hemoglobin than was possible by intermittent blood transfusion. The treatment was highly effective, but because of the fairly short half-life of the molecule at approximately 6 to 8 h, injections usually had to be administered two to three times weekly. A second-generation erythropoietin analogue, darbepoetin alfa, was then created, with a longer elimination half-life in vivo that translated into less frequent dosing, usually once weekly or once every 2 wk. More recently, another erythropoietin-related molecule has been produced called Continuous Erythropoietin Receptor Activator with an even greater half-life, and other molecules are in development or are being licensed, including biosimilar epoetin products and Hematide. The latter is a synthetic peptide-based erythropoietin receptor agonist that, interestingly, has no structural homology with erythropoietin, and yet is still able to activate the erythropoietin receptor and stimulate erythropoiesis. The search goes on for orally active antianemic therapies, and several strategies are being investigated, although none is imminently available. This article reviews the latest progress with these novel erythropoietic agents in this new era in anemia management. Clin J Am Soc Nephrol 3: 200–207, 2008. doi: 10.2215/CJN.03840907 any nephrologists can still recall the days when EPO antibodies), storage and stability (must be stored at tem- large numbers of dialysis patients were transfusion peratures of approximately 4°C), and administration (all cur- M dependent, requiring repeated red cell transfusions rently licensed products are administered intravenously or every few weeks to increase the hemoglobin concentration from subcutaneously). -

Self-Administered Specialty Drug List
Self‐Administered Drug List BRAND NAME GENERIC DRUG NME ABIRATERONE ACETATE ABIRATERONE ACETATE ACTEMRA TOCILIZUMAB ACTEMRA ACTPEN TOCILIZUMAB ACTHAR CORTICOTROPIN ACTIMMUNE INTERFERON GAMMA‐1B,RECOMB ADCIRCA TADALAFIL ADEMPAS RIOCIGUAT AFINITOR EVEROLIMUS AFINITOR DISPERZ EVEROLIMUS ALECENSA ALECTINIB HCL ALUNBRIG BRIGATINIB ALYQ TADALAFIL AMBRISENTAN AMBRISENTAN AMPYRA DALFAMPRIDINE APOKYN APOMORPHINE HCL ARANESP DARBEPOETIN ALFA IN POLYSORBAT ARCALYST RILONACEPT ARIKAYCE AMIKACIN LIPOSOMAL/NEB.ACCESSR ARIXTRA FONDAPARINUX SODIUM AUBAGIO TERIFLUNOMIDE AUSTEDO DEUTETRABENAZINE AVEED TESTOSTERONE UNDECANOATE AVONEX INTERFERON BETA‐1A AVONEX INTERFERON BETA‐1A/ALBUMIN AVONEX PEN INTERFERON BETA‐1A AYVAKIT AVAPRITINIB BAFIERTAM MONOMETHYL FUMARATE BALVERSA ERDAFITINIB BENLYSTA BELIMUMAB BETASERON INTERFERON BETA‐1B BETHKIS TOBRAMYCIN BOSENTAN BOSENTAN BOSULIF BOSUTINIB BRAFTOVI ENCORAFENIB BRONCHITOL MANNITOL BRUKINSA ZANUBRUTINIB BYNFEZIA OCTREOTIDE ACETATE CABENUVA CABOTEGRAVIR/RILPIVIRINE CABLIVI CAPLACIZUMAB‐YHDP CABOMETYX CABOZANTINIB S‐MALATE CALQUENCE ACALABRUTINIB CAPECITABINE CAPECITABINE CAPRELSA VANDETANIB CARBAGLU CARGLUMIC ACID CAYSTON AZTREONAM LYSINE CERDELGA ELIGLUSTAT TARTRATE CETROTIDE CETRORELIX ACETATE CHENODAL CHENODIOL CHOLBAM CHOLIC ACID CHORIONIC GONADOTROPIN CHORIONIC GONADOTROPIN, HUMAN CIMZIA CERTOLIZUMAB PEGOL COPAXONE GLATIRAMER ACETATE COPIKTRA DUVELISIB COSENTYX (2 SYRINGES) SECUKINUMAB COSENTYX PEN SECUKINUMAB COSENTYX PEN (2 PENS) SECUKINUMAB COSENTYX SYRINGE SECUKINUMAB COTELLIC COBIMETINIB FUMARATE CYSTADANE -

NEW 2018 Specialty Drugs
Specialty Medications by Disease Type updated 7/2/18 ALLERGY AND ASTHMA Brand Name Generic FASENRA BENRALIZUMAB NUCALA MEPOLIZUMAB XOLAIR OMALIZUMAB CYSTIC FIBROSIS Brand name Generic BETHKIS TOBRAMYCIN CAYSTON AZTREONAM CREON PANCRELIPASE HYPERSAL SODIUM CHLORIDE 7% (HYPERTONIC SALINE) KALYDECO IVACAFTOR ORKAMBI LUMACAFTOR/IVACAFTOR PANCREAZE PANCRELIPASE PERTZYE PANCRELIPASE PULMOZYME DORNASE ALFA SYMDEKO TEZACAFTOR/IVACAFTOR TOBI/ TOBI PODHALER TOBRAMYCIN ULTRESA PANCRELIPASE VIOKASE PANCRELIPASE ZENPEP PANCRELIPASE DERMATOLOGY Brand name Generic COSENTYX SECUKINUMAB DUPIXENT DUPILUMAB ENBREL ETANERCEPT HUMIRA ADALIMUMAB OTEZLA APREMILAST OTREXUP METHOTREXATE RASUVO METHOTREXATE STELARA USTEKINUMAB TALTZ IXEKIZUMAB TREMFYA GUSELKUMAB XOLAIR OMALIZUMAB ENDOCRINE DISORDERS AVEED TESTOSTERONE UNDECANOATE LUPRON LEUPROLIDE MAKENA HYDROXYPROGESTERONE CAPROATE SOMATULINE LANREOTIDE SANDOSTATIN OCTREOTIDE SUPPRELIN HISTRELIN VANTAS HISTRELIN GROWTH HORMONE DEFICIENCY Brand name Generic GENOTROPIN SOMATROPIN HUMATROPE SOMATROPIN NORDITROPIN SOMATROPIN NUTROPIN SOMATROPIN OMNITROPE SOMATROPIN SAIZEN SOMATROPIN SEROSTIM SOMATROPIN ZOMACTON SOMATROPIN ZORBTIVE SOMATROPIN HEPATITIS Brand name Generic BARACLUDE ENTECAVIR DAKLINZA DACLATASVIR EPCLUSA SOFOSBUVIR/VELPATASVIR HARVONI LEDIPASVIR/SOFOSBUVIR MAVYRET GLECAPREVIR/PIBRENTASVIR OLYSIO SIMEPREVIR PEGASYS PEGINTERFERON ALFA-2A PEGINTRON PEGINTERFERON ALFA-2B RIBAVIRIN SOVALDI SOFOSBUVIR TECHNIVIE OMBITASVIR/PARITAPREVIR/RITONAVIR VIEKIRA OMBITASVIR/PARITAPREVIR/RITONAVIR/DASABUVIR VEMLIDY TENOFOVIR