Epstein–Barr Virus Transcription Factor Zta Acts Through Distal Regulatory
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Methylation Patterns of RASA3 Associated with Clinicopathological
Journal of Cancer 2018, Vol. 9 2116 Ivyspring International Publisher Journal of Cancer 2018; 9(12): 2116-2122. doi: 10.7150/jca.24567 Research Paper Methylation patterns of RASA3 associated with clinicopathological factors in hepatocellular carcinoma Hui Lin*1,2, Xiaoxiao Fan*1,2, LiFeng He1,2, Daizhan Zhou1,2,3,4 1. Department of General Surgery, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, China 2. Biomedical Research Center, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, China. 3. Bio-X Center, Key Laboratory for the Genetics of Developmental and Neuropsychiatric Disorders (Ministry of Education), Shanghai Jiao Tong University, Shanghai, China. 4. Present address: Key Laboratory of Arrhythmias of the Ministry of Education of China, East Hospital, Tongji University School of Medicine; Institute of Medical Genetics, Tongji University, Shanghai, China. * Hui Lin and Xiaoxiao Fan contributed equally for this work. Corresponding author: Daizhan Zhou, Phd, Biomedical Research Center, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine. 3 East Qingchun Rd, Hangzhou 310016, China. Tel: 86-13738055489; Fax: 86-571-86044817; Email: [email protected]/[email protected] © Ivyspring International Publisher. This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions. Received: 2017.12.26; Accepted: 2018.03.31; Published: 2018.05.25 Abstract Hepatocellular carcinoma (HCC) is the sixth most common tumor worldwide. The relationship between the gene methylation accumulation and HCC has been widely studied. -

Effect of Cpg Sites on Transcription Factor in Promoter of 'RASA3' Gene
International Journal of Pharmaceutical and Phytopharmacological Research (eIJPPR) | February 2021 | Volume 11| Issue 1 | Page 120-124 Khloud Algothmi, Effect of CpG Sites Methylation on Transcription Factor Promoter of (RASA3) Gene Effect of CpG Sites on Transcription Factor in Promoter of ‘RASA3’ Gene Khloud Algothmi¹, Hadeel Kalboneh1, Aisha Alrofaidi¹, Mona Alharbi¹, Reem Farsi¹, Najla Alburae1, Magdah Ganash1, Alawiah Alhebshi1, Fatemah Basingab¹, Sheren Azhari1, Manal Shaabad2, Ashraf Dallol2, Amany Alqosaibi3, Mohammed Jan4, Hesham Aldhalaan5, Safiah Alhazmi¹ 1 Biological Sciences Department, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia. ² King Abdulaziz University, Center of Excellence in Genomic Medicine Research, Jeddah, Saudi Arabia. 3 Department of Biology, College of Science, Imam Abdulrahman bin Faisal University, Dammam, Saudi Arabia. 4 King Abdulaziz University, College of Medicine, Jeddah, Saudi Arabia. ⁵Center for Autism Research at King Faisal Specialist Hospital and Research Center (KFSH&RC), Saudi Arabia. ABSTRACT RAS p21 activator 3 gene is a member of the Ras GAP1 family of GTPase-activating proteins. DNA methylation is the most well-known epigenetic mechanism. Abnormality on RAS proteins which are regulated by RASA3 may play a critical role in Autism. Autism is a complex of neurodevelopmental disorders. Defining the regulators of RASA3 is important to understand its role in Autism. This study included 19 Saudi autistic children and their healthy siblings. Blood was used to analyze the differential expression of RASA3 and its transcriptional factor Specificity protein 1. qRT-PCR and MethyLight assay was used to measure CpG sites methylation on TFBs in the promoter of RASA3 to investigate the association between methylation level and TF in the regulation of RASA3 expression. -

Delineation of Yet Unknown Cryptic Subtelomere Aberrations in 50% of Acute Myeloid Leukemia with Normal GTG-Banding Karyotype
417-423 31/12/2008 08:43 Ì ™ÂÏ›‰·417 INTERNATIONAL JOURNAL OF ONCOLOGY 34: 417-423, 2009 417 Delineation of yet unknown cryptic subtelomere aberrations in 50% of acute myeloid leukemia with normal GTG-banding karyotype MADELEINE GROSS1, HASMIK MKRTCHYAN1, MELANIE GLASER1, HANS JÖRG FRICKE2, KLAUS HÖFFKEN2, ANITA HELLER1, ANJA WEISE1 and THOMAS LIEHR1 1Institut für Humangenetik und Anthropologie, Kollegiengasse 10, D-07743 Jena; 2Klinik fuer Innere Medizin II, Erlanger Allee 101, D-07747 Jena, Germany Received August 29, 2008; Accepted November 3, 2008 DOI: 10.3892/ijo_00000165 Abstract. Acute myeloid leukemia (AML) is a heterogeneous hematopoietic progenitor cells. At the time of diagnosis ~55% disease with respect to clinical prognosis and acquired of the patients show at least one chromosomal aberration in chromosomal aberrations. After routine banding cytogenetic their bone marrow blasts where >200 different aberrations are analysis 45% of AML patients show a normal karyotype described until now (1). However, 45% of the patients with (NK-AML). For a better understanding of development and de novo AML still show a cytogenetic normal karyotype progression in AML, it is important to find markers which (NK-AML) (2,3). Recent studies gave evidence that in could be primary genetic aberrations. Therefore, in this addition to chromosomal aberrations genetic changes can study 31 patients with NK-AML were analyzed by new occur. These include e.g. mutations in the FLT3 (4), the high resolution molecular cytogenetic approaches. A NPM1 (5,6) or the MLL gene (7,8). Also acquired isodisomy combination of multitude multicolor banding and metaphase were described (9-11). -

Homozygous Deletion Mapping in Myeloma Samples Identifies Genes and an Expression Signature Relevant to Pathogenesis and Outcome
Published OnlineFirst March 9, 2010; DOI: 10.1158/1078-0432.CCR-09-2831 Published Online First on March 9, 2010 as 10.1158/1078-0432.CCR-09-2831 Clinical Imaging, Diagnosis, Prognosis Cancer Research Homozygous Deletion Mapping in Myeloma Samples Identifies Genes and an Expression Signature Relevant to Pathogenesis and Outcome Nicholas J. Dickens1, Brian A. Walker1, Paola E. Leone1, David C. Johnson1, José L. Brito1, Athanasia Zeisig1, Matthew W. Jenner1, Kevin D. Boyd1, David Gonzalez1, Walter M. Gregory2, Fiona M. Ross3, Faith E. Davies1, and Gareth J. Morgan1 Abstract Purpose: Myeloma is a clonal malignancy of plasma cells. Poor-prognosis risk is currently identified by clinical and cytogenetic features. However, these indicators do not capture all prognostic information. Gene expression analysis can be used to identify poor-prognosis patients and this can be improved by combination with information about DNA-level changes. Experimental Design: Using single nucleotide polymorphism–based gene mapping in combination with global gene expression analysis, we have identified homozygous deletions in genes and networks that are relevant to myeloma pathogenesis and outcome. Results: We identified 170 genes with homozygous deletions and corresponding loss of expression. Deletion within the “cell death” network was overrepresented and cases with these deletions had im- paired overall survival. From further analysis of these events, we have generated an expression-based sig- nature associated with shorter survival in 258 patients and confirmed this signature in data from two independent groups totaling 800 patients. We defined a gene expression signature of 97 cell death genes that reflects prognosis and confirmed this in two independent data sets. -

A Role for Bivalent Genes in Epithelial to Mesenchymal Transition
A Role for Bivalent Genes in Epithelial to Mesenchymal Transition Francisco Sadras (B.Sc, Hns) Thesis submitted for the degree of Doctor of Philosophy Faculty of Sciences School of Biological Sciences Department of Molecular & Cellular Biology Affiliated with Gene Regulation Laboratory Centre for Cancer Biology IMVS, SA Pathology April 2017 Contents Abstract ...................................................................................................................................... 5 Declaration ................................................................................................................................. 8 Preface........................................................................................................................................ 9 Acknowledgments.................................................................................................................... 10 List of Figures .......................................................................................................................... 12 List of Tables ........................................................................................................................... 13 Abbreviations ........................................................................................................................... 14 Chapter 1 Introduction ........................................................................................................ 16 1.1 Preamble ................................................................................................................... -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

And Post-Synaptic Abnormalities in Schizophrenia Lynsey S
bioRxiv preprint doi: https://doi.org/10.1101/384560; this version posted August 3, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. A Transcriptome Wide Association Study implicates specific pre- and post-synaptic abnormalities in Schizophrenia Lynsey S Hall (PhD)1*, Christopher W Medway (PhD)1*, Antonio F Pardinas (PhD)1, Elliott G Rees (PhD)1, Valentina Escott-Price (PhD)1, Andrew Pocklington (PhD)1, Peter A Holmans (PhD)1, James TR Walters (MRCPsych, PhD)1, Michael J Owen (FRCPsych, PhD)1, Michael C O’Donovan (FRCPsych, PhD)1 *joint first author 1. MRC Centre for Neuropsychiatric Genetics and Genomics, Division of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK Corresponding author: Dr Lynsey Hall MRC Centre for Neuropsychiatric Genetics and Genomics Cardiff University Hadyn Ellis Building Cardiff CF24 4HQ Phone: +44 (0)29 2068 8422 Email: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/384560; this version posted August 3, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Abstract Schizophrenia is a complex highly heritable disorder. Genome-wide association studies have identified multiple loci that influence the risk of developing schizophrenia, although the causal variants driving these associations and their impacts on specific genes are largely unknown. -
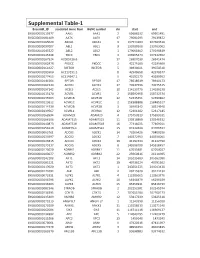
Supplemental Table-1
Supplemental Table-1 Ensembl_ID Associated Gene Nam HGNC symbol chr start end ENSG00000115977 AAK1 AAK1 2 69688532 69901481 ENSG00000181409 AATK AATK 17 79091095 79139872 ENSG00000165029 ABCA1 ABCA1 9 107543283 107690518 ENSG00000097007 ABL1 ABL1 9 133589268 133763062 ENSG00000143322 ABL2 ABL2 1 179068462 179198819 ENSG00000145348 TBCK TBCK 4 106965474 107242652 ENSG00000167524 AC005726.6 17 26907030 26941474 ENSG00000162878 PKDCC PKDCC 2 42275160 42285668 ENSG00000164327 RICTOR RICTOR 5 38938021 39074510 ENSG00000185900 AC113191.1 8 42948658 42978577 ENSG00000177453 AC114947.1 5 43192173 43280952 ENSG00000141564 RPTOR RPTOR 17 78518619 78940173 ENSG00000161533 ACOX1 ACOX1 17 73937596 73975515 ENSG00000197142 ACSL5 ACSL5 10 114133776 114188138 ENSG00000115170 ACVR1 ACVR1 2 158592958 158732374 ENSG00000135503 ACVR1B ACVR1B 12 52345451 52390862 ENSG00000123612 ACVR1C ACVR1C 2 158388888 158485517 ENSG00000114739 ACVR2B ACVR2B 3 38495342 38524948 ENSG00000139567 ACVRL1 ACVRL1 12 52301202 52317145 ENSG00000168594 ADAM29 ADAM29 4 175750819 175899331 ENSG00000166106 ADAMTS15 ADAMTS15 11 130318869 130346532 ENSG00000140873 ADAMTS18 ADAMTS18 16 77316025 77469011 ENSG00000156218 ADAMTSL3 ADAMTSL3 15 84322838 84708594 ENSG00000063761 ADCK1 ADCK1 14 78266426 78400296 ENSG00000133597 ADCK2 ADCK2 7 140372953 140396061 ENSG00000123815 ADCK4 ADCK4 19 41197434 41222790 ENSG00000173137 ADCK5 ADCK5 8 145596790 145618457 ENSG00000173020 ADRBK1 ADRBK1 11 67033881 67054027 ENSG00000100077 ADRBK2 ADRBK2 22 25960816 26118985 ENSG00000142208 AKT1 AKT1 14 105235689 -

Cutting the Brakes on Ras—Cytoplasmic Gaps As Targets of Inactivation in Cancer
cancers Review Cutting the Brakes on Ras—Cytoplasmic GAPs as Targets of Inactivation in Cancer Arianna Bellazzo and Licio Collavin * Department of Life Sciences, University of Trieste, Via L. Giorgieri 1, 34127 Trieste, Italy; [email protected] * Correspondence: [email protected] Received: 28 September 2020; Accepted: 15 October 2020; Published: 21 October 2020 Simple Summary: GTPase-Activating Proteins (RasGAPs) are a group of structurally related proteins with a fundamental role in controlling the activity of Ras in normal and cancer cells. In particular, loss of function of RasGAPs may contribute to aberrant Ras activation in cancer. Here we review the multiple molecular mechanisms and factors that are involved in downregulating RasGAPs expression and functions in cancer. Additionally, we discuss how extracellular stimuli from the tumor microenvironment can control RasGAPs expression and activity in cancer cells and stromal cells, indirectly affecting Ras activation, with implications for cancer development and progression. Abstract: The Ras pathway is frequently deregulated in cancer, actively contributing to tumor development and progression. Oncogenic activation of the Ras pathway is commonly due to point mutation of one of the three Ras genes, which occurs in almost one third of human cancers. In the absence of Ras mutation, the pathway is frequently activated by alternative means, including the loss of function of Ras inhibitors. Among Ras inhibitors, the GTPase-Activating Proteins (RasGAPs) are major players, given their ability to modulate multiple cancer-related pathways. In fact, most RasGAPs also have a multi-domain structure that allows them to act as scaffold or adaptor proteins, affecting additional oncogenic cascades. In cancer cells, various mechanisms can cause the loss of function of Ras inhibitors; here, we review the available evidence of RasGAP inactivation in cancer, with a specific focus on the mechanisms. -

Cancer Driver&Ndash
Oncogene (2014) 33, 814–822 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc ORIGINAL ARTICLE Cancer driver–passenger distinction via sporadic human and dog cancer comparison: a proof-of-principle study with colorectal cancer J Tang1,YLi1, K Lyon1, J Camps2, S Dalton1, T Ried2 and S Zhao1 Herein we report a proof-of-principle study illustrating a novel dog–human comparison strategy that addresses a central aim of cancer research, namely cancer driver–passenger distinction. We previously demonstrated that sporadic canine colorectal cancers (CRCs) share similar molecular pathogenesis mechanisms as their human counterparts. In this study, we compared the genome- wide copy number abnormalities between 29 human and 10 canine sporadic CRCs. This led to the identification of 73 driver candidate genes (DCGs), altered in both species, and with 27 from the whole genome and 46 from dog–human genomic rearrangement breakpoint (GRB) regions, as well as 38 passenger candidate genes (PCGs), altered in humans only and located in GRB regions. We noted that DCGs significantly differ from PCGs in every analysis conducted to assess their cancer relevance and biological functions. Importantly, although PCGs are not enriched in any specific functions, DCGs possess significantly enhanced functionality closely associated with cell proliferation and death regulation, as well as with epithelial cell apicobasal polarity establishment/maintenance. These observations support the notion that, in sporadic CRCs of both species, cell polarity genes not only contribute in preventing cancer cell invasion and spreading, but also likely serve as tumor suppressors by modulating cell growth. This pilot study validates our novel strategy and has uncovered four new potential cell polarity and colorectal tumor suppressor genes (RASA3, NUPL1, DENND5A and AVL9). -

Epigenomic Promoter Alterations Amplify Gene Isoform and Immunogenic Diversity in Gastric Adenocarcinoma
Published OnlineFirst March 20, 2017; DOI: 10.1158/2159-8290.CD-16-1022 RESEARCH ARTICLE Epigenomic Promoter Alterations Amplify Gene Isoform and Immunogenic Diversity in Gastric Adenocarcinoma Aditi Qamra1,2, Manjie Xing3,4, Nisha Padmanabhan3, Jeffrey Jun Ting Kwok5, Shenli Zhang3, Chang Xu3, Yan Shan Leong6, Ai Ping Lee Lim1, Qianqao Tang7, Wen Fong Ooi1, Joyce Suling Lin1, Tannistha Nandi1, Xiaosai Yao1, Xuewen Ong3, Minghui Lee3, Su Ting Tay3, Angie Tan Lay Keng3, Erna Gondo Santoso7, Cedric Chuan Young Ng7, Alvin Ng3,4, Apinya Jusakul3, Duane Smoot8, Hassan Ashktorab9, Sun Young Rha10, Khay Guan Yeoh11,12, Wei Peng Yong13, Pierce K.H. Chow14,15, Weng Hoong Chan16, Hock Soo Ong16, Khee Chee Soo15, Kyoung-Mee Kim17, Wai Keong Wong16, Steven G. Rozen3,18, Bin Tean Teh3,6,7,18, Dennis Kappei6, Jeeyun Lee19, John Connolly5,20, and Patrick Tan1,3,6,18,21 ABSTRACT Promoter elements play important roles in isoform and cell type–specific expression. We surveyed the epigenomic promoter landscape of gastric adenocarcinoma, analyz- ing 110 chromatin profiles (H3K4me3, H3K4me1, H3K27ac) of primary gastric cancers, gastric cancer lines, and nonmalignant gastric tissues. We identified nearly 2,000 promoter alterations (somatic promot- ers), many deregulated in various epithelial malignancies and mapping frequently to alternative promoters within the same gene, generating potential pro-oncogenic isoforms (RASA3). Somatic promoter– associated N-terminal peptides displaying relative depletion in tumors exhibited high-affinity MHC bind- ing predictions and elicited potent T-cell responses in vitro, suggesting a mechanism for reducing tumor antigenicity. In multiple patient cohorts, gastric cancers with high somatic promoter usage also displayed reduced T-cell cytolytic marker expression. -

Copy Number Variations in a Cohort of 420 Individuals with Neurodevelopmental Disorders from the South of Brazil
www.nature.com/scientificreports OPEN Copy Number Variations in a Cohort of 420 Individuals with Neurodevelopmental Disorders From the South of Brazil Tiago Fernando Chaves 1,5*, Nathacha Baretto1,5, Luan Freitas de Oliveira1, Maristela Ocampos2, Ingrid Tremel Barbato2, Mayara Anselmi1, Gisele Rozone De Luca3, Jorge Humberto Barbato Filho2, Louise Lapagesse de Camargo Pinto3, Pricila Bernardi4 & Angelica Francesca Maris1* Chromosomal microarray (CMA) is now recommended as frst tier for the evaluation in individuals with unexplained neurodevelopmental disorders (ND). However, in developing countries such as Brazil, classical cytogenetic tests are still the most used in clinical practice, as refected by the scarcity of publications of microarray investigation in larger cohorts. This is a retrospective study which analyses the reading fles of CMA and available clinical data from 420 patients from the south of Brazil, mostly children, with neurodevelopmental disorders requested by medical geneticists and neurologists for diagnostic purpose. Previous karyotyping was reported for 138 and includes 17 with abnormal results. The platforms used for CMA were CYTOSCAN 750K (75%) and CYTOSCAN HD (25%). The sex ratio of the patients was 1.625 males :1 female and the mean age was 9.5 years. A total of 96 pathogenic copy number variations (CNVs), 58 deletions and 38 duplications, were found in 18% of the patients and in all chromosomes, except chromosome 11. For 12% of the patients only variants of uncertain clinical signifcance were found. No clinically relevant CNV was found in 70%. The main referrals for chromosomal microarrays (CMA) were developmental delay (DD), intellectual disability (ID), facial dysmorphism and autism spectrum disorder (ASD).