Inter-Population Differences in Retrogene Loss and Expression in Humans
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PDF Datasheet
Product Datasheet C9orf85 Overexpression Lysate NBL1-08604 Unit Size: 0.1 mg Store at -80C. Avoid freeze-thaw cycles. Protocols, Publications, Related Products, Reviews, Research Tools and Images at: www.novusbio.com/NBL1-08604 Updated 3/17/2020 v.20.1 Earn rewards for product reviews and publications. Submit a publication at www.novusbio.com/publications Submit a review at www.novusbio.com/reviews/destination/NBL1-08604 Page 1 of 2 v.20.1 Updated 3/17/2020 NBL1-08604 C9orf85 Overexpression Lysate Product Information Unit Size 0.1 mg Concentration The exact concentration of the protein of interest cannot be determined for overexpression lysates. Please contact technical support for more information. Storage Store at -80C. Avoid freeze-thaw cycles. Buffer RIPA buffer Target Molecular Weight 18.1 kDa Product Description Description Transient overexpression lysate of chromosome 9 open reading frame 85 (C9orf85) The lysate was created in HEK293T cells, using Plasmid ID RC208807 and based on accession number NM_182505. The protein contains a C- MYC/DDK Tag. Gene ID 138241 Gene Symbol C9ORF85 Species Human Notes HEK293T cells in 10-cm dishes were transiently transfected with a non-lipid polymer transfection reagent specially designed and manufactured for large volume DNA transfection. Transfected cells were cultured for 48hrs before collection. The cells were lysed in modified RIPA buffer (25mM Tris-HCl pH7.6, 150mM NaCl, 1% NP-40, 1mM EDTA, 1xProteinase inhibitor cocktail mix, 1mM PMSF and 1mM Na3VO4, and then centrifuged to clarify the lysate. Protein concentration was measured by BCA protein assay kit.This product is manufactured by and sold under license from OriGene Technologies and its use is limited solely for research purposes. -

Dual Proteome-Scale Networks Reveal Cell-Specific Remodeling of the Human Interactome
bioRxiv preprint doi: https://doi.org/10.1101/2020.01.19.905109; this version posted January 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Dual Proteome-scale Networks Reveal Cell-specific Remodeling of the Human Interactome Edward L. Huttlin1*, Raphael J. Bruckner1,3, Jose Navarrete-Perea1, Joe R. Cannon1,4, Kurt Baltier1,5, Fana Gebreab1, Melanie P. Gygi1, Alexandra Thornock1, Gabriela Zarraga1,6, Stanley Tam1,7, John Szpyt1, Alexandra Panov1, Hannah Parzen1,8, Sipei Fu1, Arvene Golbazi1, Eila Maenpaa1, Keegan Stricker1, Sanjukta Guha Thakurta1, Ramin Rad1, Joshua Pan2, David P. Nusinow1, Joao A. Paulo1, Devin K. Schweppe1, Laura Pontano Vaites1, J. Wade Harper1*, Steven P. Gygi1*# 1Department of Cell Biology, Harvard Medical School, Boston, MA, 02115, USA. 2Broad Institute, Cambridge, MA, 02142, USA. 3Present address: ICCB-Longwood Screening Facility, Harvard Medical School, Boston, MA, 02115, USA. 4Present address: Merck, West Point, PA, 19486, USA. 5Present address: IQ Proteomics, Cambridge, MA, 02139, USA. 6Present address: Vor Biopharma, Cambridge, MA, 02142, USA. 7Present address: Rubius Therapeutics, Cambridge, MA, 02139, USA. 8Present address: RPS North America, South Kingstown, RI, 02879, USA. *Correspondence: [email protected] (E.L.H.), [email protected] (J.W.H.), [email protected] (S.P.G.) #Lead Contact: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.01.19.905109; this version posted January 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

Comprehensive Identification of Genes Driven by ERV9-Ltrs Reveals TNFRSF10B As a Re-Activatable Mediator of Testicular Cancer Cell Death
Cell Death and Differentiation (2016) 23, 64–75 & 2016 Macmillan Publishers Limited All rights reserved 1350-9047/16 www.nature.com/cdd Comprehensive identification of genes driven by ERV9-LTRs reveals TNFRSF10B as a re-activatable mediator of testicular cancer cell death U Beyer1,2,5, SK Krönung1,5, A Leha3, L Walter4 and M Dobbelstein*,1 The long terminal repeat (LTR) of human endogenous retrovirus type 9 (ERV9) acts as a germline-specific promoter that induces the expression of a proapoptotic isoform of the tumor suppressor homologue p63, GTAp63, in male germline cells. Testicular cancer cells silence this promoter, but inhibitors of histone deacetylases (HDACs) restore GTAp63 expression and give rise to apoptosis. We show here that numerous additional transcripts throughout the genome are driven by related ERV9-LTRs. 3' Rapid amplification of cDNA ends (3’RACE) was combined with next-generation sequencing to establish a large set of such mRNAs. HDAC inhibitors induce these ERV9-LTR-driven genes but not the LTRs from other ERVs. In particular, a transcript encoding the death receptor DR5 originates from an ERV9-LTR inserted upstream of the protein coding regions of the TNFRSF10B gene, and it shows an expression pattern similar to GTAp63. When treating testicular cancer cells with HDAC inhibitors as well as the death ligand TNF-related apoptosis-inducing ligand (TRAIL), rapid cell death was observed, which depended on TNFRSF10B expression. HDAC inhibitors also cooperate with cisplatin (cDDP) to promote apoptosis in testicular cancer cells. ERV9-LTRs not only drive a large set of human transcripts, but a subset of them acts in a proapoptotic manner. -

Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications
Stem Cell Rev and Rep DOI 10.1007/s12015-016-9662-8 Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications Behnam Ahmadian Baghbaderani 1 & Adhikarla Syama2 & Renuka Sivapatham3 & Ying Pei4 & Odity Mukherjee2 & Thomas Fellner1 & Xianmin Zeng3,4 & Mahendra S. Rao5,6 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract We have recently described manufacturing of hu- help determine which set of tests will be most useful in mon- man induced pluripotent stem cells (iPSC) master cell banks itoring the cells and establishing criteria for discarding a line. (MCB) generated by a clinically compliant process using cord blood as a starting material (Baghbaderani et al. in Stem Cell Keywords Induced pluripotent stem cells . Embryonic stem Reports, 5(4), 647–659, 2015). In this manuscript, we de- cells . Manufacturing . cGMP . Consent . Markers scribe the detailed characterization of the two iPSC clones generated using this process, including whole genome se- quencing (WGS), microarray, and comparative genomic hy- Introduction bridization (aCGH) single nucleotide polymorphism (SNP) analysis. We compare their profiles with a proposed calibra- Induced pluripotent stem cells (iPSCs) are akin to embryonic tion material and with a reporter subclone and lines made by a stem cells (ESC) [2] in their developmental potential, but dif- similar process from different donors. We believe that iPSCs fer from ESC in the starting cell used and the requirement of a are likely to be used to make multiple clinical products. We set of proteins to induce pluripotency [3]. Although function- further believe that the lines used as input material will be used ally identical, iPSCs may differ from ESC in subtle ways, at different sites and, given their immortal status, will be used including in their epigenetic profile, exposure to the environ- for many years or even decades. -

Genome-Wide Profiling of Druggable Active Tumor Defense Mechanisms to Enhance Cancer Immunotherapy
bioRxiv preprint doi: https://doi.org/10.1101/843185; this version posted November 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Genome-wide profiling of druggable active tumor defense mechanisms to enhance cancer immunotherapy Rigel J. Kishton1,2,*,#, Shashank J. Patel1,2,†,*, Suman K. Vodnala1,2, Amy E. Decker3, Yogin Patel1,2, Madhusudhanan Sukumar1,2, Tori N. Yamamoto1,2,4, Zhiya Yu1,2, Michelle Ji1,2, Amanda N. Henning1,2, Devikala Gurusamy1,2, Douglas C. Palmer1,2, Winifred Lo1, Anna Pasetto1, Parisa Malekzadeh1, Drew C. Deniger1, Kris C. Wood3, Neville E. Sanjana5,6, Nicholas P. Restifo1,2, #, § 1Surgery Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD 20892, USA 2Center for Cell-Based Therapy, National Cancer Institute, Bethesda, MD 20892, USA 3Department of Pharmacology & Cancer Biology, Duke University School of Medicine, Durham, NC, USA 4Immunology Graduate Group, University of Pennsylvania, Philadelphia, PA 19104, USA 5New York Genome Center, New York, NY 10013 USA 6Department of Biology, New York University, New York, NY 10003, USA *These authors contributed equally to this work. †Present address: NextCure Inc., Beltsville, MD 20705, USA §Present address: Lyell Immunopharma, South San Francisco, CA 94080, USA #Corresponding authors. NPR: [email protected]. RJK: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/843185; this version posted November 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. -

Wo 2010/065567 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 10 June 2010 (10.06.2010) WO 2010/065567 A2 (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 36/889 (2006.01) A61K 31/16 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 36/736 (2006.01) A61K 31/05 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, A61K 31/166 (2006.01) A61P 5/00 (2006.01) CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, PCT/US2009/066294 KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, (22) International Filing Date: ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, 1 December 2009 (01 .12.2009) NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, (25) Filing Language: English TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 61/1 18,945 1 December 2008 (01 .12.2008) US GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, (71) Applicant (for all designated States except US): LIFES¬ TM), European (AT, BE, BG, CH, CY, CZ, DE, DK, EE, PAN EXTENSION LLC [US/US]; 933 First Colonial ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, Road, Suite 114, Virginia Beach, VA 23454 (US). -

A Prevalence of Imprinted Genes Within the Total Transcriptomes of Human Tissues and Cells
Hindawi Publishing Corporation Molecular Biology International Volume 2012, Article ID 793506, 29 pages doi:10.1155/2012/793506 Research Article A Prevalence of Imprinted Genes within the Total Transcriptomes of Human Tissues and Cells Sergey V. Anisimov1, 2 1 Research Unit of Cellular and Genetic Engineering, V. A. Almazov Federal Center for Heart, Blood & Endocrinology, Akkuratova Street 2, Saint-Petersburg 197341, Russia 2 Department of Intracellular Signaling and Transport, Institute of Cytology, Russian Academy of Sciences, Tikhoretsky Prosp. 4, Saint-Petersburg 194064, Russia Correspondence should be addressed to Sergey V. Anisimov, [email protected] Received 14 March 2012; Revised 23 June 2012; Accepted 28 June 2012 Academic Editor: Alessandro Desideri Copyright © 2012 Sergey V. Anisimov. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Genomic imprinting is an epigenetic phenomenon that causes a differential expression of paternally and maternally inherited alleles of a subset of genes (the so-called imprinted genes). Imprinted genes are distributed throughout the genome and it is predicted that about 1% of the human genes may be imprinted. It is recognized that the allelic expression of imprinted genes varies between tissues and developmental stages. The current study represents the first attempt to estimate a prevalence of imprinted genes within the total human transcriptome. In silico analysis of the normalized expression profiles of a comprehensive panel of 173 established and candidate human imprinted genes was performed, in 492 publicly available SAGE libraries. The latter represent human cell and tissue samples in a variety of physiological and pathological conditions. -

Nearly All New Protein-Coding Predictions in the CHESS Database Are Not Protein-Coding Irwin Jungreis*,✝,1,2, Michael L
bioRxiv preprint doi: https://doi.org/10.1101/360602; this version posted July 2, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Nearly all new protein-coding predictions in the CHESS database are not protein-coding Irwin Jungreis*,✝,1,2, Michael L. Tress*,3, Jonathan Mudge*,4, Cristina Sisu5,6, Toby Hunt4, Rory Johnson7,8, Barbara Uszczynska-Ratajczak9, Julien Lagarde10,11,12, James Wright13, Paul Muir14,15, Mark Gerstein5,16,17, Roderic Guigo10,11,12, Manolis Kellis1,2, Adam Frankish✝,4, Paul Flicek4, The GENCODE Consortium 1MIT Computer Science and Artificial Intelligence Laboratory, Cambridge, MA; 2Broad Institute of MIT and Harvard, Cambridge, MA; 3Bioinformatics Unit, Spanish National Cancer Research Centre, Madrid, Spain; 4European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, UK; 5Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520, USA; 6Department of Bioscience, Brunel University London, Uxbridge, UB8 3PH, UK; 7Department of Medical Oncology, Inselspital, University Hospital, University of Bern, Bern, Switzerland; 8Department of Biomedical Research (DBMR), University of Bern, Bern, Switzerland; 9Centre of New Technologies, University of Warsaw, Warsaw, Poland; 10Centre for Genomic Regulation (CRG), The Barcelona Institute for -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

A Prevalence of Imprinted Genes Within the Total Transcriptomes of Human Tissues and Cells
Hindawi Publishing Corporation Molecular Biology International Volume 2012, Article ID 793506, 29 pages doi:10.1155/2012/793506 Research Article A Prevalence of Imprinted Genes within the Total Transcriptomes of Human Tissues and Cells Sergey V. Anisimov1, 2 1 Research Unit of Cellular and Genetic Engineering, V. A. Almazov Federal Center for Heart, Blood & Endocrinology, Akkuratova Street 2, Saint-Petersburg 197341, Russia 2 Department of Intracellular Signaling and Transport, Institute of Cytology, Russian Academy of Sciences, Tikhoretsky Prosp. 4, Saint-Petersburg 194064, Russia Correspondence should be addressed to Sergey V. Anisimov, [email protected] Received 14 March 2012; Revised 23 June 2012; Accepted 28 June 2012 Academic Editor: Alessandro Desideri Copyright © 2012 Sergey V. Anisimov. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Genomic imprinting is an epigenetic phenomenon that causes a differential expression of paternally and maternally inherited alleles of a subset of genes (the so-called imprinted genes). Imprinted genes are distributed throughout the genome and it is predicted that about 1% of the human genes may be imprinted. It is recognized that the allelic expression of imprinted genes varies between tissues and developmental stages. The current study represents the first attempt to estimate a prevalence of imprinted genes within the total human transcriptome. In silico analysis of the normalized expression profiles of a comprehensive panel of 173 established and candidate human imprinted genes was performed, in 492 publicly available SAGE libraries. The latter represent human cell and tissue samples in a variety of physiological and pathological conditions. -
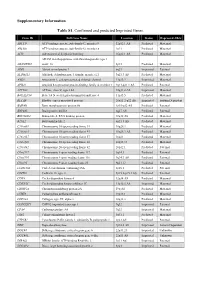
Supplementary Information Table S1. Confirmed and Predicted Imprinted
Supplementary Information Table S1. Confirmed and predicted Imprinted Genes. Gene ID Full Gene Name Location Status Expressed Allele ABCC9 ATP-binding cassette, sub-family C, member 9 12p12.1 AS Predicted Maternal ABCG8 ATP-binding cassette, sub-family G, member 8 2p21 Predicted Maternal ACD Adrenocortical dysplasia homolog 16q22.1 AS Predicted Maternal ADAM metallopeptidase with thrombospondin type 1 ADAMTS16 motif, 16 5p15 Predicted Maternal AIM1 Absent in melanoma 1 6q21 Imprinted Paternal ALDH1L1 Aldehyde dehydrogenase 1 family, member L1 3q21.3 AS Predicted Maternal ANO1 Anoctamin 1, calcium activated chloride channel 11q13.3 Imprinted Maternal APBA1 Amyloid beta precursor protein-binding, family A, member 1 9q13-q21.1 AS Predicted Paternal ATP10A ATPase, class V, type 10A 15q11.2 AS Imprinted Maternal B4GALNT4 Beta-1,4-N-acetyl-galactosaminyl transferase 4 11p15.5 Predicted Maternal BLCAP Bladder cancer associated protein 20q11.2-q12 AS Imprinted Isoform Dependent BMP8B Bone morphogenetic protein 8b 1p35-p32 AS Predicted Paternal BRP44L Brain protein 44-like 6q27 AS Predicted Paternal BRUNOL4 Bruno-like 4, RNA binding protein 18q12 AS Predicted Maternal BTNL2 Butyrophilin-like 2 6p21.3 AS Predicted Maternal C10orf91 Chromosome 10 open reading frame 91 10q26.3 Predicted Maternal C10orf93 Chromosome 10 open reading frame 93 10q26.3 AS Predicted Maternal C16orf57 Chromosome 16 open reading frame 57 16q21 Predicted Maternal C20orf20 Chromosome 20 open reading frame 20 20q13.33 Predicted Maternal C20orf82 Chromosome 20 open reading frame