Building a Reference Transcriptome for Juniperus Squamata (Cupressaceae) Based on Single-Molecule Real-Time Sequencing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phylogenetic Analyses of Juniperus Species in Turkey and Their Relations with Other Juniperus Based on Cpdna Supervisor: Prof
MOLECULAR PHYLOGENETIC ANALYSES OF JUNIPERUS L. SPECIES IN TURKEY AND THEIR RELATIONS WITH OTHER JUNIPERS BASED ON cpDNA A THESIS SUBMITTED TO THE GRADUATE SCHOOL OF NATURAL AND APPLIED SCIENCES OF MIDDLE EAST TECHNICAL UNIVERSITY BY AYSUN DEMET GÜVENDİREN IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN BIOLOGY APRIL 2015 Approval of the thesis MOLECULAR PHYLOGENETIC ANALYSES OF JUNIPERUS L. SPECIES IN TURKEY AND THEIR RELATIONS WITH OTHER JUNIPERS BASED ON cpDNA submitted by AYSUN DEMET GÜVENDİREN in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Department of Biological Sciences, Middle East Technical University by, Prof. Dr. Gülbin Dural Ünver Dean, Graduate School of Natural and Applied Sciences Prof. Dr. Orhan Adalı Head of the Department, Biological Sciences Prof. Dr. Zeki Kaya Supervisor, Dept. of Biological Sciences METU Examining Committee Members Prof. Dr. Musa Doğan Dept. Biological Sciences, METU Prof. Dr. Zeki Kaya Dept. Biological Sciences, METU Prof.Dr. Hayri Duman Biology Dept., Gazi University Prof. Dr. İrfan Kandemir Biology Dept., Ankara University Assoc. Prof. Dr. Sertaç Önde Dept. Biological Sciences, METU Date: iii I hereby declare that all information in this document has been obtained and presented in accordance with academic rules and ethical conduct. I also declare that, as required by these rules and conduct, I have fully cited and referenced all material and results that are not original to this work. Name, Last name : Aysun Demet GÜVENDİREN Signature : iv ABSTRACT MOLECULAR PHYLOGENETIC ANALYSES OF JUNIPERUS L. SPECIES IN TURKEY AND THEIR RELATIONS WITH OTHER JUNIPERS BASED ON cpDNA Güvendiren, Aysun Demet Ph.D., Department of Biological Sciences Supervisor: Prof. -

Morphology and Morphogenesis of the Seed Cones of the Cupressaceae - Part II Cupressoideae
1 2 Bull. CCP 4 (2): 51-78. (10.2015) A. Jagel & V.M. Dörken Morphology and morphogenesis of the seed cones of the Cupressaceae - part II Cupressoideae Summary The cone morphology of the Cupressoideae genera Calocedrus, Thuja, Thujopsis, Chamaecyparis, Fokienia, Platycladus, Microbiota, Tetraclinis, Cupressus and Juniperus are presented in young stages, at pollination time as well as at maturity. Typical cone diagrams were drawn for each genus. In contrast to the taxodiaceous Cupressaceae, in Cupressoideae outgrowths of the seed-scale do not exist; the seed scale is completely reduced to the ovules, inserted in the axil of the cone scale. The cone scale represents the bract scale and is not a bract- /seed scale complex as is often postulated. Especially within the strongly derived groups of the Cupressoideae an increased number of ovules and the appearance of more than one row of ovules occurs. The ovules in a row develop centripetally. Each row represents one of ascending accessory shoots. Within a cone the ovules develop from proximal to distal. Within the Cupressoideae a distinct tendency can be observed shifting the fertile zone in distal parts of the cone by reducing sterile elements. In some of the most derived taxa the ovules are no longer (only) inserted axillary, but (additionally) terminal at the end of the cone axis or they alternate to the terminal cone scales (Microbiota, Tetraclinis, Juniperus). Such non-axillary ovules could be regarded as derived from axillary ones (Microbiota) or they develop directly from the apical meristem and represent elements of a terminal short-shoot (Tetraclinis, Juniperus). -
Botanical Name Common Name
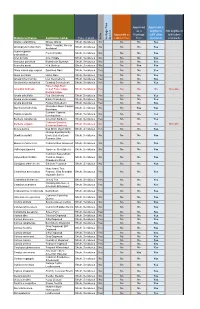
Approved Approved & as a eligible to Not eligible to Approved as Frontage fulfill other fulfill other Type of plant a Street Tree Tree standards standards Heritage Tree Tree Heritage Species Botanical Name Common name Native Abelia x grandiflora Glossy Abelia Shrub, Deciduous No No No Yes White Forsytha; Korean Abeliophyllum distichum Shrub, Deciduous No No No Yes Abelialeaf Acanthropanax Fiveleaf Aralia Shrub, Deciduous No No No Yes sieboldianus Acer ginnala Amur Maple Shrub, Deciduous No No No Yes Aesculus parviflora Bottlebrush Buckeye Shrub, Deciduous No No No Yes Aesculus pavia Red Buckeye Shrub, Deciduous No No Yes Yes Alnus incana ssp. rugosa Speckled Alder Shrub, Deciduous Yes No No Yes Alnus serrulata Hazel Alder Shrub, Deciduous Yes No No Yes Amelanchier humilis Low Serviceberry Shrub, Deciduous Yes No No Yes Amelanchier stolonifera Running Serviceberry Shrub, Deciduous Yes No No Yes False Indigo Bush; Amorpha fruticosa Desert False Indigo; Shrub, Deciduous Yes No No No Not eligible Bastard Indigo Aronia arbutifolia Red Chokeberry Shrub, Deciduous Yes No No Yes Aronia melanocarpa Black Chokeberry Shrub, Deciduous Yes No No Yes Aronia prunifolia Purple Chokeberry Shrub, Deciduous Yes No No Yes Groundsel-Bush; Eastern Baccharis halimifolia Shrub, Deciduous No No Yes Yes Baccharis Summer Cypress; Bassia scoparia Shrub, Deciduous No No No Yes Burning-Bush Berberis canadensis American Barberry Shrub, Deciduous Yes No No Yes Common Barberry; Berberis vulgaris Shrub, Deciduous No No No No Not eligible European Barberry Betula pumila -

Abiotic Factors and Yushania Influences on Abies Forest Composition in Taiwan
Taiwania, 59(3): 247‒261, 2014 DOI: 10.6165/tai.2014.59.247 RESEARCH ARTICLE Abiotic Factors and Yushania Influences on Abies Forest Composition in Taiwan Cheng-Tao Lin(1), Tzu-Ying Chen(2), Chang-Fu Hsieh(3) and Chyi-Rong Chiou(1*) 1. School of Forestry and Resource Conservation, National Taiwan University, No. 1, Sect. 4, Roosevelt Rd., Taipei, 10617, Taiwan. 2. Department of Forestry and Natural Resources, National Ilan University, Sect. 1, Shen-Lung Rd., Ilan, 26047, Taiwan. 3. Institute of Ecology and Evolutionary Biology, National Taiwan University, No. 1, Sect. 4, Roosevelt Rd., Taipei, 10617, Taiwan. * Corresponding author. Tel.: +886-2-3366-4640; Fax: +886-2-2365-4520; Email: [email protected] (Manuscript received 20 March 2014; accepted 26 May 2014) ABSTRACT: Abies kawakamii forests are generally distributed above 3,000 m in Taiwanese high mountains. The community data used in our analysis were derived from the database of the National Vegetation Diversity Inventory and Mapping Project of Taiwan (NVDIMP), and environmental data were obtained from the WorldClim and NVDIMP databases. We used non-metric multidimensional scaling (NMDS) to identify vegetation composition of Abies communities and the structural equation models (SEMs) were used to examine the complex relationships between environmental factors and vegetation composition. The results of ordination showed the most important factors determining species composition of Abies forests involved habitat rockiness, heat load index, warmth index and summer and winter. SEM results approved the warmth index and winter precipitation were the main drivers determining the latent variable—climate, which significantly affect the overstory composition of Abies communities. -

Juniper (Juniperus Davurica ‘Parsonii’, J
Juniper (Juniperus davurica ‘Parsonii’, J. conferta 'Blue Pacific', J. squamata 'Blue Star') Tolerance to Glyphosate. Mark Andrew Czarnota Department of Horticulture - Griffin The University of Georgia NATURE OF WORK: To evaluate the tolerance of 3 juniper species to varying rates of glyphosate. For many reasons preemergent herbicides can fail to control weeds. Once weed seeds germinate, nurseryman and landscapers are often left with hand weeding as their only control option. Including junipers, many plants are known to be very tolerant to glyphosate. Moreover, glyphosate can control a wide number of herbaceous plants at 0.5 lbs. ai./A and lower. The goal of the test is to determine the maximum amount of glyphosate that can be applied without damaging select juniper species. MATERIAL AND METHODS: On July 27, 2003 at the Center for Applied Nursery Research, 18, 1 gallon pots of each of the following junipers were assambled: Shore juniper (Juniperus conferta 'Blue pacific'), Dahurian juniper (J. davurica 'Parsoni'), Singleseed juniper (J. squamata Blue Star'). Nine, one gallon pots, of each species were placed in a 6 ft. x 6 ft. area. Herbicide treatments were applied to the area containing the 27 one gallon pots. Pots were then carefully moved to an evaluation area were they were arranged in a randomized complete block (RCB) design. Each treatment contained 3 replications, and each replication contained 3 subsamples. The process was continued for each herbicide treatment. Sprays were applied with a CO2 backpack sprayer calibrated to deliver 20 gallons per acer (GPA). Watering occurred on an as needed basis, and this represented approximately ½ to 1 inch of water per day. -

Vol 5.35 March/April 2017
March & April 2017 Vol. 5 No. 35 Proud to sponsor the Yarra Proud sponsor of the Yarra Valley Bonsai Society Valley Bonsai Society www.facebook.com/BonsaiRoots2015 www.bonsaisensation.com.au The Yarra Valley Bonsai Society Newsletter PO Box 345 Mount Evelyn, VIC 3796 Australia www.yarravalleybonsai.org.au Highlights from March & April 2017 [email protected] Reg. Assoc A0052264P Our March club meeting featured Trevor McComb from Bonsai Art who spoke and demonstrated Pres: Geoff P 0430 130 955 on Japanese Shimpaku junipers (Juniperus chinensis var. Sargentii). Sec: Lindsay H 0403 800 671 Junipers are a large family comprising 60 - 70 varieties, native to Europe, Asia, Africa, and North Treas: Julie H 0419 870 240 Marktg: Marlene J 0418 369 852 America. None are indigenous to Australia. They are long-lived, some up to 1000 years old, and range from ground covers to trees that may be 30m tall. Juniperus squamata is the most common The YVBS meets on the juniper available locally due to its fast second Tuesday of each growth rate being more attractive to the month now at Cire Services (Formerly Upper Yarra Com- nursery industry, but shimpaku is 10 munity House or Morrison times better for bonsai purposes. Shim- House) paku‟s advantages include hardiness, Old Hereford Road, long-lived, flexible, less dieback, softer Mount Evelyn foliage, and drought tolerant. It is also Meetings begin at 7:30pm . possible to get back-budding on old 118 B8 wood. Shimpaku are often shown with the bark Saturday Workshops are 2- 4:30pm on the Last Saturday rubbed off. -

Evergreens.Pdf
Buxus - Boxwood Green Mountain Boxwood Buxus ‘Green Mountain’ Height: 3 - 4 Feet (1.25 m) Spread: 3 Feet (0.9 m) Foliage: Green Hardiness: Zone 4 The most upright boxwood, with dark green foliage year round. Slow growing, rounded plant. Chamaecyparis - Falsecypress Golden Mops Threadleaf Falsecypress Chamaecyparis pisifera ‘Filifera Mops’ Height: 3 Feet (1 m) Spread: 3 Feet (1 m) Green Mountain Boxwood Shape: Mound Hardiness: Zone 4 Soft, feathery, thread-like branches that are a striking yellow in colour. A slow growing plant that forms an irregular mound. Juniperus - Spreading Juniper Arcadia Juniper Juniperus sabina ‘Arcadia’ Height: 24-30 Inches (0.75m) Spread: 3 - 5 Feet (1.5 m) Foliage: Medium green Hardiness: Zone 2 This all purpose juniper has soft foliage that is a medium green in colour. The branch habit is semi- spreading. Prefers full sun. Arcadia Juniper Blue Chip Juniper Juniperus horizontalis ‘Blue Chip’ EVERGREENS & CONIFERS EVERGREENS Height: 9 - 12 Inches (0.3 m) Spread: 3 - 5 Feet (1.5 m) & CONIFERS EVERGREENS Foliage: Silver blue Hardiness: Zone 3 A low mounding juniper with branches radiating from the centre. The silver blue colour of the foli- age is retained throughout the year. Blue Danube Juniper Juniperus sabina ‘Blue Danube’ Height: 24-30 Inches (0.8 m) Spread: 3 - 5 Feet (1.5 m) Foliage: Blue green Hardiness: Zone 3 A broad, semi-erect and spreading plant with shimmering, blue-green foliage. Blue Chip Juniper Blue Forest Juniper Juniperus sabina ‘Blue Forest’ Height: 12 Inches (0.3 m) Spread: 3 - 5 Feet (1.5 m) Foliage: Blue green Hardiness: Zone 3 A low growing plant with erect branches so spread is reduced. -

Traverse Mountain Approved Plant List
08.03.2010 Traverse Mountain Approved Plant List Evergreen Trees Type Scientific Name Common Name Sun/Shade Water Req. Zone Color Evergreen Trees Abies concolor White Fir Full Sun/Part Shade Medium 3 Silver Blue-Green Evergreen Trees Abies lasiocarpa 'Arizonica' Corkbark Fir Full Sun Low 4 Blue-Green Evergreen Trees Chamaecyparis nootkatensis Alaskan Cedar Full Sun Low 4 Blue-Green Evergreen Trees Cedrus Alantica Glauca Blue Atlas Cedar Full Sun Moderate 3 Silver Blue-Green Evergreen Trees Cedrus Deodora Prostratra 'Emeral Prostrate Deodora Cedar Full Sun Moderate 3 Green- Silvery Blue Evergreen Trees Juniperus osteosperma Utah Juniper Full Sun Low 4-7 Green Evergreen Trees Juniperus scopulorum Rocky Mountain Juniper Full Sun Low 2 Green Evergreen Trees Juniperus squamata 'Blue Alps' Flaky Juniper Full Sun Medium 4 Medium Blue Evergreen Trees Juniperus virginiana 'Cupressifolia' Hillspire Juniper Full Sun Low 5 Green Evergreen Trees Juniperus virginiana 'Skyrocket' Skyrocket Juniper Full Sun Low 5 Silver-gray Evergreen Trees Picea Abies Norway Spruce Full Sun-Light Shade Low 3 Dark Green Evergreen Trees Picea englemannii Englemann Spruce Full Sun Low 2 Green Evergreen Trees Picea Omorika Serbian Spruce Full Sun Low 2 Green Evergreen Trees Picea Pungens Colorado Spruce Full Sun Low 2 Green Evergreen Trees Picea Pungens Glauca Colorado Blue Spruce Full Sun Low 2 Blue Green Evergreen Trees Picea glauca densata Black Hills Spruce Full Sun Medium 3 Green Evergreen Trees Picea pungens glauca 'Baby Blue Eyes' Baby Blue Eyes Spruce Full Sun Medium -

Notes on Cultivated Junipers
Butler University Botanical Studies Volume 14 Article 7 Notes on Cultivated Junipers Marion T. Hall Follow this and additional works at: https://digitalcommons.butler.edu/botanical The Butler University Botanical Studies journal was published by the Botany Department of Butler University, Indianapolis, Indiana, from 1929 to 1964. The scientific journal eaturf ed original papers primarily on plant ecology, taxonomy, and microbiology. Recommended Citation Hall, Marion T. (1961) "Notes on Cultivated Junipers," Butler University Botanical Studies: Vol. 14 , Article 7. Retrieved from: https://digitalcommons.butler.edu/botanical/vol14/iss1/7 This Article is brought to you for free and open access by Digital Commons @ Butler University. It has been accepted for inclusion in Butler University Botanical Studies by an authorized editor of Digital Commons @ Butler University. For more information, please contact [email protected]. Butler University Botanical Studies (1929-1964) Edited by J. E. Potzger The Butler University Botanical Studies journal was published by the Botany Department of Butler University, Indianapolis, Indiana, from 1929 to 1964. The scientific journal featured original papers primarily on plant ecology, taxonomy, and microbiology. The papers contain valuable historical studies, especially floristic surveys that document Indiana’s vegetation in past decades. Authors were Butler faculty, current and former master’s degree students and undergraduates, and other Indiana botanists. The journal was started by Stanley Cain, noted conservation biologist, and edited through most of its years of production by Ray C. Friesner, Butler’s first botanist and founder of the department in 1919. The journal was distributed to learned societies and libraries through exchange. During the years of the journal’s publication, the Butler University Botany Department had an active program of research and student training. -

Alpine Vegetation of the Khangchendzonga Landscape, Sikkim Himalaya: Community Characteristics, Diversity and Aspects of Ecology
ALPINE VEGETATION OF THE KHANGCHENDZONGA LANDSCAPE, SIKKIM HIMALAYA: COMMUNITY CHARACTERISTICS, DIVERSITY AND ASPECTS OF ECOLOGY Sandeep Tambe and G. S. Rawat ABSTRACT he alpine vegetation of the Sikkim Himalaya has received limited attention despite being a part of the eastern Himalaya global biodiversity hotspot. The current study undertaken in the third highest landscape in the world - Tthe Khangchendzonga National Park (KNP), provides information on the different alpine vegetation communities and aspects of their ecology. The transverse spurs from the unique north-south running Khangchendzonga range result in a landscape level differentiation of the Outer, Inner and Tibetan Himalaya in just 50 km distance. The alpine vegetation based on numerical classification has been segregated into 11 types with the extensive ones being Juniperus indica scrub, Rhododendron scrub, Kobresia duthiei moist meadow, Kobresia nepalensis moist meadow, Kobresia pygmaea dry meadow and Anaphalis xylorhiza mixed meadow. Based on Canonical Correspondence Analysis, the three environmental gradients of rainfall, elevation, and soil were found to be the primary determinants of vegetation pattern. A total of 585 species of angiosperms belonging to 67 families and 243 genera were recorded in 390 km2 area. Compared to the alpine zone of western Himalaya, proportions of alpine scrub and sedge meadows were higher whereas herbaceous formations and grassy meadows were limited in extent. The alpha species diversity was found to be lower mainly because the alpine region -

The Evolution of Cavitation Resistance in Conifers Maximilian Larter
The evolution of cavitation resistance in conifers Maximilian Larter To cite this version: Maximilian Larter. The evolution of cavitation resistance in conifers. Bioclimatology. Univer- sit´ede Bordeaux, 2016. English. <NNT : 2016BORD0103>. <tel-01375936> HAL Id: tel-01375936 https://tel.archives-ouvertes.fr/tel-01375936 Submitted on 3 Oct 2016 HAL is a multi-disciplinary open access L'archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destin´eeau d´ep^otet `ala diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publi´esou non, lished or not. The documents may come from ´emanant des ´etablissements d'enseignement et de teaching and research institutions in France or recherche fran¸caisou ´etrangers,des laboratoires abroad, or from public or private research centers. publics ou priv´es. THESE Pour obtenir le grade de DOCTEUR DE L’UNIVERSITE DE BORDEAUX Spécialité : Ecologie évolutive, fonctionnelle et des communautés Ecole doctorale: Sciences et Environnements Evolution de la résistance à la cavitation chez les conifères The evolution of cavitation resistance in conifers Maximilian LARTER Directeur : Sylvain DELZON (DR INRA) Co-Directeur : Jean-Christophe DOMEC (Professeur, BSA) Soutenue le 22/07/2016 Devant le jury composé de : Rapporteurs : Mme Amy ZANNE, Prof., George Washington University Mr Jordi MARTINEZ VILALTA, Prof., Universitat Autonoma de Barcelona Examinateurs : Mme Lisa WINGATE, CR INRA, UMR ISPA, Bordeaux Mr Jérôme CHAVE, DR CNRS, UMR EDB, Toulouse i ii Abstract Title: The evolution of cavitation resistance in conifers Abstract Forests worldwide are at increased risk of widespread mortality due to intense drought under current and future climate change. -

Cone Morphology in Juniperus in the Light of Cone Evolution in Cupressaceae S.L
Flora (2003) 198, 161–177 http://www.urbanfischer.de/journals/flora Cone morphology in Juniperus in the light of cone evolution in Cupressaceae s.l. Christian Schulz, Armin Jagel & Thomas Stützel* Lehrstuhl für Spezielle Botanik, Ruhr-University Bochum, Universitätsstr. 150, D-44780 Bochum, Germany Submitted: Nov 28, 2002 · Accepted: Jan 23, 2003 Summary The interpretation of the berry-like, fleshy cones of Juniperus was up to now based on concepts about the conifer cone which are dismissed since Florin (1951). Comparative morphological and developmental studies showed that ovules alternating with the last whorl of cone scales cannot be regarded as part of a sporophyll (cone scale; pre-Florin interpretation). These alternating ovules are inserted directly on the cone axis and continue the phyllotactic pattern of the cone scales. If the usual seed scale is regarded as an axillary brachyblast (short-shoot) bearing ovules, the ovules alternating with ultimate whorls of cone scales in Juniperus sect. Juniperus can be regarded as a brachyblast terminating the cone axis. This interpretation allows to establish a standard bauplan for Cupressaceae in which species of Cupressus and Juniperus form a transition series towards more and more reduced cones. This series coincides with phylogenetic trees based on molecular studies. Key words: Juniperus, Cupressaceae, cone, morphogenesis, ovule, evolution Introduction should have led to a phylogenetic secondary shift of the remaining ovule into the gap between the scales. Herz- The fleshy, berry-like cones of Juniperus communis L. feld (1914) differs from this interpretation in assuming and other species of Juniperus sect. Juniperus (= sect. an ontogenetic shift (fig.