Genasm: a High-Performance, Low-Power Approximate String Matching Acceleration Framework for Genome Sequence Analysis Damla Senol Cali†On Gurpreet S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Problem of Fuzzy Duplicate Detection of Large Texts
The problem of fuzzy duplicate detection of large texts E V Sharapova1 and R V Sharapov1 1Vladimir State University, Orlovskaya street 23, Murom, Russia, 602264 Abstract. In the paper, considered the problem of fuzzy duplicate detection. There are given the basic approaches to detection of text duplicates–distance between strings, fuzzy search algorithms without indexing data, fuzzy search algorithms with indexing data. Thereview of existing methods for the fuzzy duplicate detection is given. The algorithm of fuzzy duplicate detection is present. The algorithm of fuzzy duplicate texts detection was implemented in the system AVTOR.NET. The paper presents the results of testing of the system.The use of filtering text, stemming and character replacement, allow the algorithm to found duplicates even in minor modified texts. 1. Introduction Today computer technology and the World Wide Web have become part of our lives. Billions of users use the Internet every day. The Internet contains a huge number of documents. Many books are transformed in a digital form. Users can read books, articles, newspapers directly from computers and electronic gadgets. Today, in the Internet there are many studying materials: lectures, textbooks, methodical literature, etc. There are big collections of essays, course and diploma projects and scientific dissertations. It leads to problem un-controlling coping of information. The text of document may be a part of another document, formed from several documents, may be modified part of another document (with a change of words to synonyms, endings, times, etc.). In this way, the document is fuzzy duplicate of other documents. There is the problem of text checking to presence of fussy duplicates. -

Comparative Analysis of Multiple Sequence Alignment Tools
I.J. Information Technology and Computer Science, 2018, 8, 24-30 Published Online August 2018 in MECS (http://www.mecs-press.org/) DOI: 10.5815/ijitcs.2018.08.04 Comparative Analysis of Multiple Sequence Alignment Tools Eman M. Mohamed Faculty of Computers and Information, Menoufia University, Egypt E-mail: [email protected]. Hamdy M. Mousa, Arabi E. keshk Faculty of Computers and Information, Menoufia University, Egypt E-mail: [email protected], [email protected]. Received: 24 April 2018; Accepted: 07 July 2018; Published: 08 August 2018 Abstract—The perfect alignment between three or more global alignment algorithm built-in dynamic sequences of Protein, RNA or DNA is a very difficult programming technique [1]. This algorithm maximizes task in bioinformatics. There are many techniques for the number of amino acid matches and minimizes the alignment multiple sequences. Many techniques number of required gaps to finds globally optimal maximize speed and do not concern with the accuracy of alignment. Local alignments are more useful for aligning the resulting alignment. Likewise, many techniques sub-regions of the sequences, whereas local alignment maximize accuracy and do not concern with the speed. maximizes sub-regions similarity alignment. One of the Reducing memory and execution time requirements and most known of Local alignment is Smith-Waterman increasing the accuracy of multiple sequence alignment algorithm [2]. on large-scale datasets are the vital goal of any technique. The paper introduces the comparative analysis of the Table 1. Pairwise vs. multiple sequence alignment most well-known programs (CLUSTAL-OMEGA, PSA MSA MAFFT, BROBCONS, KALIGN, RETALIGN, and Compare two biological Compare more than two MUSCLE). -

Influence of Angular Velocity of Pedaling on the Accuracy of The
Research article 2018-04-10 - Rev08 Influence of Angular Velocity of Pedaling on the Accuracy of the Measurement of Cyclist Power Abstract Almost all cycling power meters currently available on the The miscalculation may be—even significantly—greater than we market are positioned on rotating parts of the bicycle (pedals, found in our study, for the following reasons: crank arms, spider, bottom bracket/hub) and, regardless of • the test was limited to only 5 cyclists: there is no technical and construction differences, all calculate power on doubt other cyclists may have styles of pedaling with the basis of two physical quantities: torque and angular velocity greater variations of angular velocity; (or rotational speed – cadence). Both these measures vary only 2 indoor trainer models were considered: other during the 360 degrees of each revolution. • models may produce greater errors; The torque / force value is usually measured many times during slopes greater than 5% (the only value tested) may each rotation, while the angular velocity variation is commonly • lead to less uniform rotations and consequently neglected, considering only its average value for each greater errors. revolution (cadence). It should be noted that the error observed in this analysis This, however, introduces an unpredictable error into the power occurs because to measure power the power meter considers calculation. To use the average value of angular velocity means the average angular velocity of each rotation. In power meters to consider each pedal revolution as perfectly smooth and that use this type of calculation, this error must therefore be uniform: but this type of pedal revolution does not exist in added to the accuracy stated by the manufacturer. -

Algorithms for Approximate String Matching Petro Protsyk (28 October 2016) Agenda
Algorithms for approximate string matching Petro Protsyk (28 October 2016) Agenda • About ZyLAB • Overview • Algorithms • Brute-force recursive Algorithm • Wagner and Fischer Algorithm • Algorithms based on Automaton • Bitap • Q&A About ZyLAB ZyLAB is a software company headquartered in Amsterdam. In 1983 ZyLAB was the first company providing a full-text search program for MS-DOS called ZyINDEX In 1991 ZyLAB released ZyIMAGE software bundle that included a fuzzy string search algorithm to overcome scanning and OCR errors. Currently ZyLAB develops eDiscovery and Information Governance solutions for On Premises, SaaS and Cloud installations. ZyLAB continues to develop new versions of its proprietary full-text search engine. 3 About ZyLAB • We develop for Windows environments only, including Azure • Software developed in C++, .Net, SQL • Typical clients are from law enforcement, intelligence, fraud investigators, law firms, regulators etc. • FTC (Federal Trade Commission), est. >50 TB (peak 2TB per day) • US White House (all email of presidential administration), est. >20 TB • Dutch National Police, est. >20 TB 4 ZyLAB Search Engine Full-text search engine • Boolean and Proximity search operators • Support for Wildcards, Regular Expressions and Fuzzy search • Support for numeric and date searches • Search in document fields • Near duplicate search 5 ZyLAB Search Engine • Highly parallel indexing and searching • Can index Terabytes of data and millions of documents • Can be distributed • Supports 100+ languages, Unicode characters 6 Overview Approximate string matching or fuzzy search is the technique of finding strings in text or dictionary that match given pattern approximately with respect to chosen edit distance. The edit distance is the number of primitive operations necessary to convert the string into an exact match. -

Basement Flood Mitigation
1 Mitigation refers to measures taken now to reduce losses in the future. How can homeowners and renters protect themselves and their property from a devastating loss? 2 There are a range of possible causes for basement flooding and some potential remedies. Many of these low-cost options can be factored into a family’s budget and accomplished over the several months that precede storm season. 3 There are four ways water gets into your basement: Through the drainage system, known as the sump. Backing up through the sewer lines under the house. Seeping through cracks in the walls and floor. Through windows and doors, called overland flooding. 4 Gutters can play a huge role in keeping basements dry and foundations stable. Water damage caused by clogged gutters can be severe. Install gutters and downspouts. Repair them as the need arises. Keep them free of debris. 5 Channel and disperse water away from the home by lengthening the run of downspouts with rigid or flexible extensions. Prevent interior intrusion through windows and replace weather stripping as needed. 6 Many varieties of sturdy window well covers are available, simple to install and hinged for easy access. Wells should be constructed with gravel bottoms to promote drainage. Remove organic growth to permit sunlight and ventilation. 7 Berms and barriers can help water slope away from the home. The berm’s slope should be about 1 inch per foot and extend for at least 10 feet. It is important to note permits are required any time a homeowner alters the elevation of the property. -
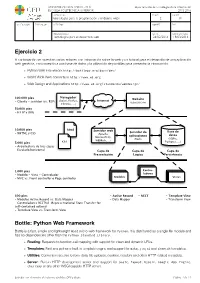
Ejercicio 2 Bottle: Python Web Framework
UNIVERSIDAD SAN PABLO - CEU departamento de tecnologías de la información ESCUELA POLITÉCNICA SUPERIOR 2015-2016 ASIGNATURA CURSO GRUPO tecnologías para la programación y el diseño web i 2 01 CALIFICACION EVALUACION APELLIDOS NOMBRE DNI OBSERVACIONES FECHA FECHA ENTREGA Tecnologías para el desarrollo web 24/02/2016 18/03/2016 Ejercicio 2 A continuación se muestran varios enlaces con información sobre la web y un tutorial para el desarrollo de una aplicación web genérica, con conexión a una base de datos y la utilización de plantillas para presentar la información. ‣ Python Web Framework http://bottlepy.org/docs/dev/ ‣ World Wide Web consortium http://www.w3.org ‣ Web Design and Applications http://www.w3.org/standards/webdesign/ Navegador 100.000 pies Website (Safari, Firefox, Internet - Cliente - servidor (vs. P2P) uspceu.com Chrome, ...) 50.000 pies - HTTP y URIs 10.000 pies html Servidor web Servidor de Base de - XHTML y CSS (Apache, aplicaciones datos Microsoft IIS, (Rack) (SQlite, WEBRick, ...) 5.000 pies css Postgres, ...) - Arquitectura de tres capas - Escalado horizontal Capa de Capa de Capa de Presentación Lógica Persistencia 1.000 pies Contro- - Modelo - Vista - Controlador ladores - MVC vs. Front controller o Page controller Modelos Vistas 500 pies - Active Record - REST - Template View - Modelos Active Record vs. Data Mapper - Data Mapper - Transform View - Controladores RESTful (Representational State Transfer for self-contained actions) - Template View vs. Transform View Bottle: Python Web Framework Bottle is a fast, simple and lightweight WSGI micro web-framework for Python. It is distributed as a single file module and has no dependencies other than the Python Standard Library. -

Simple Harmonic Motion
[SHIVOK SP211] October 30, 2015 CH 15 Simple Harmonic Motion I. Oscillatory motion A. Motion which is periodic in time, that is, motion that repeats itself in time. B. Examples: 1. Power line oscillates when the wind blows past it 2. Earthquake oscillations move buildings C. Sometimes the oscillations are so severe, that the system exhibiting oscillations break apart. 1. Tacoma Narrows Bridge Collapse "Gallopin' Gertie" a) http://www.youtube.com/watch?v=j‐zczJXSxnw II. Simple Harmonic Motion A. http://www.youtube.com/watch?v=__2YND93ofE Watch the video in your spare time. This professor is my teaching Idol. B. In the figure below snapshots of a simple oscillatory system is shown. A particle repeatedly moves back and forth about the point x=0. Page 1 [SHIVOK SP211] October 30, 2015 C. The time taken for one complete oscillation is the period, T. In the time of one T, the system travels from x=+x , to –x , and then back to m m its original position x . m D. The velocity vector arrows are scaled to indicate the magnitude of the speed of the system at different times. At x=±x , the velocity is m zero. E. Frequency of oscillation is the number of oscillations that are completed in each second. 1. The symbol for frequency is f, and the SI unit is the hertz (abbreviated as Hz). 2. It follows that F. Any motion that repeats itself is periodic or harmonic. G. If the motion is a sinusoidal function of time, it is called simple harmonic motion (SHM). -

Hub City Powertorque® Shaft Mount Reducers
Hub City PowerTorque® Shaft Mount Reducers PowerTorque® Features and Description .................................................. G-2 PowerTorque Nomenclature ............................................................................................ G-4 Selection Instructions ................................................................................ G-5 Selection By Horsepower .......................................................................... G-7 Mechanical Ratings .................................................................................... G-12 ® Shaft Mount Reducers Dimensions ................................................................................................ G-14 Accessories ................................................................................................ G-15 Screw Conveyor Accessories ..................................................................... G-22 G For Additional Models of Shaft Mount Reducers See Hub City Engineering Manual Sections F & J DOWNLOAD AVAILABLE CAD MODELS AT: WWW.HUBCITYINC.COM Certified prints are available upon request EMAIL: [email protected] • www.hubcityinc.com G-1 Hub City PowerTorque® Shaft Mount Reducers Ten models available from 1/4 HP through 200 HP capacity Manufacturing Quality Manufactured to the highest quality 98.5% standards in the industry, assembled Efficiency using precision manufactured components made from top quality per Gear Stage! materials Designed for the toughest applications in the industry Housings High strength ductile -

WEB2PY Enterprise Web Framework (2Nd Edition)
WEB2PY Enterprise Web Framework / 2nd Ed. Massimo Di Pierro Copyright ©2009 by Massimo Di Pierro. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning, or otherwise, except as permitted under Section 107 or 108 of the 1976 United States Copyright Act, without either the prior written permission of the Publisher, or authorization through payment of the appropriate per-copy fee to the Copyright Clearance Center, Inc., 222 Rosewood Drive, Danvers, MA 01923, (978) 750-8400, fax (978) 646-8600, or on the web at www.copyright.com. Requests to the Copyright owner for permission should be addressed to: Massimo Di Pierro School of Computing DePaul University 243 S Wabash Ave Chicago, IL 60604 (USA) Email: [email protected] Limit of Liability/Disclaimer of Warranty: While the publisher and author have used their best efforts in preparing this book, they make no representations or warranties with respect to the accuracy or completeness of the contents of this book and specifically disclaim any implied warranties of merchantability or fitness for a particular purpose. No warranty may be created ore extended by sales representatives or written sales materials. The advice and strategies contained herein may not be suitable for your situation. You should consult with a professional where appropriate. Neither the publisher nor author shall be liable for any loss of profit or any other commercial damages, including but not limited to special, incidental, consequential, or other damages. Library of Congress Cataloging-in-Publication Data: WEB2PY: Enterprise Web Framework Printed in the United States of America. -

"Phylogenetic Analysis of Protein Sequence Data Using The
Phylogenetic Analysis of Protein Sequence UNIT 19.11 Data Using the Randomized Axelerated Maximum Likelihood (RAXML) Program Antonis Rokas1 1Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee ABSTRACT Phylogenetic analysis is the study of evolutionary relationships among molecules, phenotypes, and organisms. In the context of protein sequence data, phylogenetic analysis is one of the cornerstones of comparative sequence analysis and has many applications in the study of protein evolution and function. This unit provides a brief review of the principles of phylogenetic analysis and describes several different standard phylogenetic analyses of protein sequence data using the RAXML (Randomized Axelerated Maximum Likelihood) Program. Curr. Protoc. Mol. Biol. 96:19.11.1-19.11.14. C 2011 by John Wiley & Sons, Inc. Keywords: molecular evolution r bootstrap r multiple sequence alignment r amino acid substitution matrix r evolutionary relationship r systematics INTRODUCTION the baboon-colobus monkey lineage almost Phylogenetic analysis is a standard and es- 25 million years ago, whereas baboons and sential tool in any molecular biologist’s bioin- colobus monkeys diverged less than 15 mil- formatics toolkit that, in the context of pro- lion years ago (Sterner et al., 2006). Clearly, tein sequence analysis, enables us to study degree of sequence similarity does not equate the evolutionary history and change of pro- with degree of evolutionary relationship. teins and their function. Such analysis is es- A typical phylogenetic analysis of protein sential to understanding major evolutionary sequence data involves five distinct steps: (a) questions, such as the origins and history of data collection, (b) inference of homology, (c) macromolecules, developmental mechanisms, sequence alignment, (d) alignment trimming, phenotypes, and life itself. -

Performance Evaluation of Leading Protein Multiple Sequence Alignment Methods
International Journal of Engineering and Advanced Technology (IJEAT) ISSN: 2249 – 8958, Volume-9 Issue-1, October 2019 Performance Evaluation of Leading Protein Multiple Sequence Alignment Methods Arunima Mishra, B. K. Tripathi, S. S. Soam MSA is a well-known method of alignment of three or more Abstract: Protein Multiple sequence alignment (MSA) is a biological sequences. Multiple sequence alignment is a very process, that helps in alignment of more than two protein intricate problem, therefore, computation of exact MSA is sequences to establish an evolutionary relationship between the only feasible for the very small number of sequences which is sequences. As part of Protein MSA, the biological sequences are not practical in real situations. Dynamic programming as used aligned in a way to identify maximum similarities. Over time the sequencing technologies are becoming more sophisticated and in pairwise sequence method is impractical for a large number hence the volume of biological data generated is increasing at an of sequences while performing MSA and therefore the enormous rate. This increase in volume of data poses a challenge heuristic algorithms with approximate approaches [7] have to the existing methods used to perform effective MSA as with the been proved more successful. Generally, various biological increase in data volume the computational complexities also sequences are organized into a two-dimensional array such increases and the speed to process decreases. The accuracy of that the residues in each column are homologous or having the MSA is another factor critically important as many bioinformatics same functionality. Many MSA methods were developed over inferences are dependent on the output of MSA. -
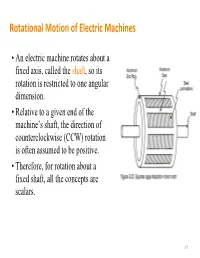
Rotational Motion of Electric Machines
Rotational Motion of Electric Machines • An electric machine rotates about a fixed axis, called the shaft, so its rotation is restricted to one angular dimension. • Relative to a given end of the machine’s shaft, the direction of counterclockwise (CCW) rotation is often assumed to be positive. • Therefore, for rotation about a fixed shaft, all the concepts are scalars. 17 Angular Position, Velocity and Acceleration • Angular position – The angle at which an object is oriented, measured from some arbitrary reference point – Unit: rad or deg – Analogy of the linear concept • Angular acceleration =d/dt of distance along a line. – The rate of change in angular • Angular velocity =d/dt velocity with respect to time – The rate of change in angular – Unit: rad/s2 position with respect to time • and >0 if the rotation is CCW – Unit: rad/s or r/min (revolutions • >0 if the absolute angular per minute or rpm for short) velocity is increasing in the CCW – Analogy of the concept of direction or decreasing in the velocity on a straight line. CW direction 18 Moment of Inertia (or Inertia) • Inertia depends on the mass and shape of the object (unit: kgm2) • A complex shape can be broken up into 2 or more of simple shapes Definition Two useful formulas mL2 m J J() RRRR22 12 3 1212 m 22 JRR()12 2 19 Torque and Change in Speed • Torque is equal to the product of the force and the perpendicular distance between the axis of rotation and the point of application of the force. T=Fr (Nm) T=0 T T=Fr • Newton’s Law of Rotation: Describes the relationship between the total torque applied to an object and its resulting angular acceleration.