HMMER User's Guide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Comparative Analysis of Multiple Sequence Alignment Tools
I.J. Information Technology and Computer Science, 2018, 8, 24-30 Published Online August 2018 in MECS (http://www.mecs-press.org/) DOI: 10.5815/ijitcs.2018.08.04 Comparative Analysis of Multiple Sequence Alignment Tools Eman M. Mohamed Faculty of Computers and Information, Menoufia University, Egypt E-mail: [email protected]. Hamdy M. Mousa, Arabi E. keshk Faculty of Computers and Information, Menoufia University, Egypt E-mail: [email protected], [email protected]. Received: 24 April 2018; Accepted: 07 July 2018; Published: 08 August 2018 Abstract—The perfect alignment between three or more global alignment algorithm built-in dynamic sequences of Protein, RNA or DNA is a very difficult programming technique [1]. This algorithm maximizes task in bioinformatics. There are many techniques for the number of amino acid matches and minimizes the alignment multiple sequences. Many techniques number of required gaps to finds globally optimal maximize speed and do not concern with the accuracy of alignment. Local alignments are more useful for aligning the resulting alignment. Likewise, many techniques sub-regions of the sequences, whereas local alignment maximize accuracy and do not concern with the speed. maximizes sub-regions similarity alignment. One of the Reducing memory and execution time requirements and most known of Local alignment is Smith-Waterman increasing the accuracy of multiple sequence alignment algorithm [2]. on large-scale datasets are the vital goal of any technique. The paper introduces the comparative analysis of the Table 1. Pairwise vs. multiple sequence alignment most well-known programs (CLUSTAL-OMEGA, PSA MSA MAFFT, BROBCONS, KALIGN, RETALIGN, and Compare two biological Compare more than two MUSCLE). -

"Phylogenetic Analysis of Protein Sequence Data Using The
Phylogenetic Analysis of Protein Sequence UNIT 19.11 Data Using the Randomized Axelerated Maximum Likelihood (RAXML) Program Antonis Rokas1 1Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee ABSTRACT Phylogenetic analysis is the study of evolutionary relationships among molecules, phenotypes, and organisms. In the context of protein sequence data, phylogenetic analysis is one of the cornerstones of comparative sequence analysis and has many applications in the study of protein evolution and function. This unit provides a brief review of the principles of phylogenetic analysis and describes several different standard phylogenetic analyses of protein sequence data using the RAXML (Randomized Axelerated Maximum Likelihood) Program. Curr. Protoc. Mol. Biol. 96:19.11.1-19.11.14. C 2011 by John Wiley & Sons, Inc. Keywords: molecular evolution r bootstrap r multiple sequence alignment r amino acid substitution matrix r evolutionary relationship r systematics INTRODUCTION the baboon-colobus monkey lineage almost Phylogenetic analysis is a standard and es- 25 million years ago, whereas baboons and sential tool in any molecular biologist’s bioin- colobus monkeys diverged less than 15 mil- formatics toolkit that, in the context of pro- lion years ago (Sterner et al., 2006). Clearly, tein sequence analysis, enables us to study degree of sequence similarity does not equate the evolutionary history and change of pro- with degree of evolutionary relationship. teins and their function. Such analysis is es- A typical phylogenetic analysis of protein sential to understanding major evolutionary sequence data involves five distinct steps: (a) questions, such as the origins and history of data collection, (b) inference of homology, (c) macromolecules, developmental mechanisms, sequence alignment, (d) alignment trimming, phenotypes, and life itself. -

Performance Evaluation of Leading Protein Multiple Sequence Alignment Methods
International Journal of Engineering and Advanced Technology (IJEAT) ISSN: 2249 – 8958, Volume-9 Issue-1, October 2019 Performance Evaluation of Leading Protein Multiple Sequence Alignment Methods Arunima Mishra, B. K. Tripathi, S. S. Soam MSA is a well-known method of alignment of three or more Abstract: Protein Multiple sequence alignment (MSA) is a biological sequences. Multiple sequence alignment is a very process, that helps in alignment of more than two protein intricate problem, therefore, computation of exact MSA is sequences to establish an evolutionary relationship between the only feasible for the very small number of sequences which is sequences. As part of Protein MSA, the biological sequences are not practical in real situations. Dynamic programming as used aligned in a way to identify maximum similarities. Over time the sequencing technologies are becoming more sophisticated and in pairwise sequence method is impractical for a large number hence the volume of biological data generated is increasing at an of sequences while performing MSA and therefore the enormous rate. This increase in volume of data poses a challenge heuristic algorithms with approximate approaches [7] have to the existing methods used to perform effective MSA as with the been proved more successful. Generally, various biological increase in data volume the computational complexities also sequences are organized into a two-dimensional array such increases and the speed to process decreases. The accuracy of that the residues in each column are homologous or having the MSA is another factor critically important as many bioinformatics same functionality. Many MSA methods were developed over inferences are dependent on the output of MSA. -

Apply Parallel Bioinformatics Applications on Linux PC Clusters
Tunghai Science Vol. : 125−141 125 July, 2003 Apply Parallel Bioinformatics Applications on Linux PC Clusters Yu-Lun Kuo and Chao-Tung Yang* Abstract In addition to the traditional massively parallel computers, distributed workstation clusters now play an important role in scientific computing perhaps due to the advent of commodity high performance processors, low-latency/high-band width networks and powerful development tools. As we know, bioinformatics tools can speed up the analysis of large-scale sequence data, especially about sequence alignment. To fully utilize the relatively inexpensive CPU cycles available to today’s scientists, a PC cluster consists of one master node and seven slave nodes (16 processors totally), is proposed and built for bioinformatics applications. We use the mpiBLAST and HMMer on parallel computer to speed up the process for sequence alignment. The mpiBLAST software uses a message-passing library called MPI (Message Passing Interface) and the HMMer software uses a software package called PVM (Parallel Virtual Machine), respectively. The system architecture and performances of the cluster are also presented in this paper. Keywords: Parallel computing, Bioinformatics, BLAST, HMMer, PC Clusters, Speedup. 1. Introduction Extraordinary technological improvements over the past few years in areas such as microprocessors, memory, buses, networks, and software have made it possible to assemble groups of inexpensive personal computers and/or workstations into a cost effective system that functions in concert and posses tremendous processing power. Cluster computing is not new, but in company with other technical capabilities, particularly in the area of networking, this class of machines is becoming a high-performance platform for parallel and distributed applications [1, 2, 11, 12, 13, 14, 15, 16, 17]. -

HMMER User's Guide
HMMER User’s Guide Biological sequence analysis using profile hidden Markov models http://hmmer.org/ Version 3.0rc1; February 2010 Sean R. Eddy for the HMMER Development Team Janelia Farm Research Campus 19700 Helix Drive Ashburn VA 20147 USA http://eddylab.org/ Copyright (C) 2010 Howard Hughes Medical Institute. Permission is granted to make and distribute verbatim copies of this manual provided the copyright notice and this permission notice are retained on all copies. HMMER is licensed and freely distributed under the GNU General Public License version 3 (GPLv3). For a copy of the License, see http://www.gnu.org/licenses/. HMMER is a trademark of the Howard Hughes Medical Institute. 1 Contents 1 Introduction 5 How to avoid reading this manual . 5 How to avoid using this software (links to similar software) . 5 What profile HMMs are . 5 Applications of profile HMMs . 6 Design goals of HMMER3 . 7 What’s still missing in HMMER3 . 8 How to learn more about profile HMMs . 9 2 Installation 10 Quick installation instructions . 10 System requirements . 10 Multithreaded parallelization for multicores is the default . 11 MPI parallelization for clusters is optional . 11 Using build directories . 12 Makefile targets . 12 3 Tutorial 13 The programs in HMMER . 13 Files used in the tutorial . 13 Searching a sequence database with a single profile HMM . 14 Step 1: build a profile HMM with hmmbuild . 14 Step 2: search the sequence database with hmmsearch . 16 Searching a profile HMM database with a query sequence . 22 Step 1: create an HMM database flatfile . 22 Step 2: compress and index the flatfile with hmmpress . -

Syntax Highlighting for Computational Biology Artem Babaian1†, Anicet Ebou2, Alyssa Fegen3, Ho Yin (Jeffrey) Kam4, German E
bioRxiv preprint doi: https://doi.org/10.1101/235820; this version posted December 20, 2017. The copyright holder has placed this preprint (which was not certified by peer review) in the Public Domain. It is no longer restricted by copyright. Anyone can legally share, reuse, remix, or adapt this material for any purpose without crediting the original authors. bioSyntax: Syntax Highlighting For Computational Biology Artem Babaian1†, Anicet Ebou2, Alyssa Fegen3, Ho Yin (Jeffrey) Kam4, German E. Novakovsky5, and Jasper Wong6. 5 10 15 Affiliations: 1. Terry Fox Laboratory, BC Cancer, Vancouver, BC, Canada. [[email protected]] 2. Departement de Formation et de Recherches Agriculture et Ressources Animales, Institut National Polytechnique Felix Houphouet-Boigny, Yamoussoukro, Côte d’Ivoire. [[email protected]] 20 3. Faculty of Science, University of British Columbia, Vancouver, BC, Canada [[email protected]] 4. Faculty of Mathematics, University of Waterloo, Waterloo, ON, Canada. [[email protected]] 5. Department of Medical Genetics, University of British Columbia, Vancouver, BC, 25 Canada. [[email protected]] 6. Genome Science and Technology, University of British Columbia, Vancouver, BC, Canada. [[email protected]] Correspondence†: 30 Artem Babaian Terry Fox Laboratory BC Cancer Research Centre 675 West 10th Avenue Vancouver, BC, Canada. V5Z 1L3. 35 Email: [[email protected]] bioRxiv preprint doi: https://doi.org/10.1101/235820; this version posted December 20, 2017. The copyright holder has placed this preprint (which was not certified by peer review) in the Public Domain. It is no longer restricted by copyright. Anyone can legally share, reuse, remix, or adapt this material for any purpose without crediting the original authors. -

Software List for Biology, Bioinformatics and Biostatistics CCT
Software List for biology, bioinformatics and biostatistics v CCT - Delta Software Version Application short read assembler and it works on both small and large (mammalian size) ALLPATHS-LG 52488 genomes provides a fast, flexible C++ API & toolkit for reading, writing, and manipulating BAMtools 2.4.0 BAM files a high level of alignment fidelity and is comparable to other mainstream Barracuda 0.7.107b alignment programs allows one to intersect, merge, count, complement, and shuffle genomic bedtools 2.25.0 intervals from multiple files Bfast 0.7.0a universal DNA sequence aligner tool analysis and comprehension of high-throughput genomic data using the R Bioconductor 3.2 statistical programming BioPython 1.66 tools for biological computation written in Python a fast approach to detecting gene-gene interactions in genome-wide case- Boost 1.54.0 control studies short read aligner geared toward quickly aligning large sets of short DNA Bowtie 1.1.2 sequences to large genomes Bowtie2 2.2.6 Bowtie + fully supports gapped alignment with affine gap penalties BWA 0.7.12 mapping low-divergent sequences against a large reference genome ClustalW 2.1 multiple sequence alignment program to align DNA and protein sequences assembles transcripts, estimates their abundances for differential expression Cufflinks 2.2.1 and regulation in RNA-Seq samples EBSEQ (R) 1.10.0 identifying genes and isoforms differentially expressed EMBOSS 6.5.7 a comprehensive set of sequence analysis programs FASTA 36.3.8b a DNA and protein sequence alignment software package FastQC -

Computational Protocol for Assembly and Analysis of SARS-Ncov-2 Genomes
PROTOCOL Computational Protocol for Assembly and Analysis of SARS-nCoV-2 Genomes Mukta Poojary1,2, Anantharaman Shantaraman1, Bani Jolly1,2 and Vinod Scaria1,2 1CSIR Institute of Genomics and Integrative Biology, Mathura Road, Delhi 2Academy of Scientific and Innovative Research (AcSIR) *Corresponding email: MP [email protected]; AS [email protected]; BJ [email protected]; VS [email protected] ABSTRACT SARS-CoV-2, the pathogen responsible for the ongoing Coronavirus Disease 2019 pandemic is a novel human-infecting strain of Betacoronavirus. The outbreak that initially emerged in Wuhan, China, rapidly spread to several countries at an alarming rate leading to severe global socio-economic disruption and thus overloading the healthcare systems. Owing to the high rate of infection of the virus, as well as the absence of vaccines or antivirals, there is a lack of robust mechanisms to control the outbreak and contain its transmission. Rapid advancement and plummeting costs of high throughput sequencing technologies has enabled sequencing of the virus in several affected individuals globally. Deciphering the viral genome has the potential to help understand the epidemiology of the disease as well as aid in the development of robust diagnostics, novel treatments and prevention strategies. Towards this effort, we have compiled a comprehensive protocol for analysis and interpretation of the sequencing data of SARS-CoV-2 using easy-to-use open source utilities. In this protocol, we have incorporated strategies to assemble the genome of SARS-CoV-2 using two approaches: reference-guided and de novo. Strategies to understand the diversity of the local strain as compared to other global strains have also been described in this protocol. -
PTIR: Predicted Tomato Interactome Resource
www.nature.com/scientificreports OPEN PTIR: Predicted Tomato Interactome Resource Junyang Yue1,*, Wei Xu1,*, Rongjun Ban2,*, Shengxiong Huang1, Min Miao1, Xiaofeng Tang1, Guoqing Liu1 & Yongsheng Liu1,3 Received: 15 October 2015 Protein-protein interactions (PPIs) are involved in almost all biological processes and form the basis Accepted: 08 April 2016 of the entire interactomics systems of living organisms. Identification and characterization of these Published: 28 April 2016 interactions are fundamental to elucidating the molecular mechanisms of signal transduction and metabolic pathways at both the cellular and systemic levels. Although a number of experimental and computational studies have been performed on model organisms, the studies exploring and investigating PPIs in tomatoes remain lacking. Here, we developed a Predicted Tomato Interactome Resource (PTIR), based on experimentally determined orthologous interactions in six model organisms. The reliability of individual PPIs was also evaluated by shared gene ontology (GO) terms, co-evolution, co-expression, co-localization and available domain-domain interactions (DDIs). Currently, the PTIR covers 357,946 non-redundant PPIs among 10,626 proteins, including 12,291 high-confidence, 226,553 medium-confidence, and 119,102 low-confidence interactions. These interactions are expected to cover 30.6% of the entire tomato proteome and possess a reasonable distribution. In addition, ten randomly selected PPIs were verified using yeast two-hybrid (Y2H) screening or a bimolecular fluorescence complementation (BiFC) assay. The PTIR was constructed and implemented as a dedicated database and is available at http://bdg.hfut.edu.cn/ptir/index.html without registration. The increasing number of complete genome sequences has revealed the entire structure and composition of proteins, based mainly on theoretical predictions utilizing their corresponding DNA sequences. -

Embnet.News Volume 4 Nr
embnet.news Volume 4 Nr. 3 Page 1 embnet.news Volume 4 Nr 3 (ISSN1023-4144) upon our core expertise in sequence analysis. Editorial Such debates can only be construed as healthy. Stasis can all too easily become stagnation. After some cliff-hanging As well as being the Christmas Bumper issue, this is also recounts and reballots at the AGM there have been changes the after EMBnet AGM Issue. The 11th Annual Business in all of EMBnet's committees. It is to be hoped that new Meeting was organised this year by the Italian Node (CNR- committee members will help galvanise us all into a more Bari) and took place up in the hills at Selva di Fasano at the active phase after a relatively quiet 1997. The fact that the end of September. For us northerners, the concept of "O for financial status of EMBnet is presently very healthy, will a beaker full of the warm south" so affected one of the certainly not impede this drive. Everyone agrees that delegates that he jumped (or was he pushed ?) fully clothed bioinformatics is one of science's growth areas and there is into the hotel swimming pool. Despite the balmy weather nobody better equipped than EMBnet to make solid and the excellent food and wine, we did manage to get a contributions to the field. solid day and a half of work done. The embnet.news editorial board: EMBnet is having to make some difficult choices about what its future direction and purpose should be. Our major source Alan Bleasby of funds is from the EU, but pretty much all countries which Rob Harper are eligible for EU funding have already joined the Robert Herzog organisation. -

Supplementary Figures
SUPPLEMENTARY FIGURES Figure S1. Read-through transcription by RNAPII in Seb1-deficient cells. (A) Average distribution of RNAPII density in wild-type (WT) and Seb1-depleted cells (Pnmt1-seb1), as determined by analysis of ChIP-seq results for all (n = 7005), mRNA (n = 5123), snoRNA (n = 53) and snRNA (n = 7) genes. Light grey rectangles represent open reading frames (mRNA genes) or noncoding sequences (snoRNA and snRNA genes) and black arrows 500 base pairs (bp) of upstream and downstream flanking DNA sequences. The absolute levels of RNAPII binding (y-axis) depend on the number of genes taken into account. Lower values for snoRNA and snRNA reflect the low number of genes present in these categories as compared to mRNA. (B) As in (A) but for (i) tandem genes that are more than 200 bp apart (n = 1425), (ii) tandem genes that are more than 400 bp apart (n = 855), (iii) convergent genes that are more than 200 bp apart (n = 452) as well as (iv) convergent genes that are more than 400 bp apart (n = 208). Figure S2. Seb1 is not the functional ortholog of Nrd1p. (A) Ten-fold serial dilutions of S. cerevisiae wild-type (WT) cells transformed with an empty vector (EV) as well as PGAL1-3XHA-NRD1 cells that were transformed with an EV or constructs that express 3XFLAG-tagged versions of S. pombe Seb1 or S. cerevisiae Nrd1. Cells were spotted on galactose- and glucose-containing media to allow and repress transcription from the PGAL1 promoter, respectively. (B) TOP: Schematic representation of the SNR13-TRS31 genomic environment. -
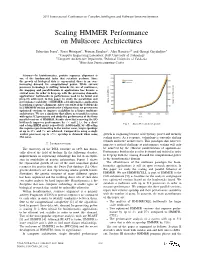
Scaling HMMER Performance on Multicore Architectures
2011 International Conference on Complex, Intelligent, and Software Intensive Systems Scaling HMMER Performance on Multicore Architectures Sebastian Isaza∗, Ernst Houtgast∗, Friman Sanchezy, Alex Ramirezyz and Georgi Gaydadjiev∗ ∗Computer Engineering Laboratory, Delft University of Technology yComputer Architecture Department, Technical University of Catalonia zBarcelona Supercomputing Center Abstract—In bioinformatics, protein sequence alignment is one of the fundamental tasks that scientists perform. Since the growth of biological data is exponential, there is an ever- increasing demand for computational power. While current processor technology is shifting towards the use of multicores, the mapping and parallelization of applications has become a critical issue. In order to keep up with the processing demands, applications’ bottlenecks to performance need to be found and properly addressed. In this paper we study the parallelism and performance scalability of HMMER, a bioinformatics application to perform sequence alignment. After our study of the bottlenecks in a HMMER version ported to the Cell processor, we present two optimized versions to improve scalability in a larger multicore architecture. We use a simulator that allows us to model a system with up to 512 processors and study the performance of the three parallel versions of HMMER. Results show that removing the I/O bottleneck improves performance by 3× and 2:4× for a short Fig. 1. Swiss-Prot database growth. and a long HMM query respectively. Additionally, by offloading the sequence pre-formatting to the worker cores, larger speedups of up to 27× and 7× are achieved. Compared to using a single worker processor, up to 156× speedup is obtained when using growth is stagnating because of frequency, power and memory 256 cores.