Open Source Python Framework for Segmentation of Biomedical Images Tobias M
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mastering Machine Learning with Scikit-Learn
www.it-ebooks.info Mastering Machine Learning with scikit-learn Apply effective learning algorithms to real-world problems using scikit-learn Gavin Hackeling BIRMINGHAM - MUMBAI www.it-ebooks.info Mastering Machine Learning with scikit-learn Copyright © 2014 Packt Publishing All rights reserved. No part of this book may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, without the prior written permission of the publisher, except in the case of brief quotations embedded in critical articles or reviews. Every effort has been made in the preparation of this book to ensure the accuracy of the information presented. However, the information contained in this book is sold without warranty, either express or implied. Neither the author, nor Packt Publishing, and its dealers and distributors will be held liable for any damages caused or alleged to be caused directly or indirectly by this book. Packt Publishing has endeavored to provide trademark information about all of the companies and products mentioned in this book by the appropriate use of capitals. However, Packt Publishing cannot guarantee the accuracy of this information. First published: October 2014 Production reference: 1221014 Published by Packt Publishing Ltd. Livery Place 35 Livery Street Birmingham B3 2PB, UK. ISBN 978-1-78398-836-5 www.packtpub.com Cover image by Amy-Lee Winfield [email protected]( ) www.it-ebooks.info Credits Author Project Coordinator Gavin Hackeling Danuta Jones Reviewers Proofreaders Fahad Arshad Simran Bhogal Sarah -

Installing Python Ø Basics of Programming • Data Types • Functions • List Ø Numpy Library Ø Pandas Library What Is Python?
Python in Data Science Programming Ha Khanh Nguyen Agenda Ø What is Python? Ø The Python Ecosystem • Installing Python Ø Basics of Programming • Data Types • Functions • List Ø NumPy Library Ø Pandas Library What is Python? • “Python is an interpreted high-level general-purpose programming language.” – Wikipedia • It supports multiple programming paradigms, including: • Structured/procedural • Object-oriented • Functional The Python Ecosystem Source: Fabien Maussion’s “Getting started with Python” Workshop Installing Python • Install Python through Anaconda/Miniconda. • This allows you to create and work in Python environments. • You can create multiple environments as needed. • Highly recommended: install Python through Miniconda. • Are we ready to “play” with Python yet? • Almost! • Most data scientists use Python through Jupyter Notebook, a web application that allows you to create a virtual notebook containing both text and code! • Python Installation tutorial: [Mac OS X] [Windows] For today’s seminar • Due to the time limitation, we will be using Google Colab instead. • Google Colab is a free Jupyter notebook environment that runs entirely in the cloud, so you can run Python code, write report in Jupyter Notebook even without installing the Python ecosystem. • It is NOT a complete alternative to installing Python on your local computer. • But it is a quick and easy way to get started/try out Python. • You will need to log in using your university account (possible for some schools) or your personal Google account. Basics of Programming Indentations – Not Braces • Other languages (R, C++, Java, etc.) use braces to structure code. # R code (not Python) a = 10 if (a < 5) { result = TRUE b = 0 } else { result = FALSE b = 100 } a = 10 if (a < 5) { result = TRUE; b = 0} else {result = FALSE; b = 100} Basics of Programming Indentations – Not Braces • Python uses whitespaces (tabs or spaces) to structure code instead. -
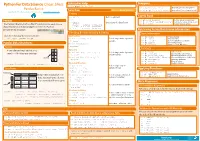
Cheat Sheet Pandas Python.Indd
Python For Data Science Cheat Sheet Asking For Help Dropping >>> help(pd.Series.loc) Pandas Basics >>> s.drop(['a', 'c']) Drop values from rows (axis=0) Selection Also see NumPy Arrays >>> df.drop('Country', axis=1) Drop values from columns(axis=1) Learn Python for Data Science Interactively at www.DataCamp.com Getting Sort & Rank >>> s['b'] Get one element -5 Pandas >>> df.sort_index() Sort by labels along an axis Get subset of a DataFrame >>> df.sort_values(by='Country') Sort by the values along an axis The Pandas library is built on NumPy and provides easy-to-use >>> df[1:] Assign ranks to entries Country Capital Population >>> df.rank() data structures and data analysis tools for the Python 1 India New Delhi 1303171035 programming language. 2 Brazil Brasília 207847528 Retrieving Series/DataFrame Information Selecting, Boolean Indexing & Setting Basic Information Use the following import convention: By Position (rows,columns) Select single value by row & >>> df.shape >>> import pandas as pd >>> df.iloc[[0],[0]] >>> df.index Describe index Describe DataFrame columns 'Belgium' column >>> df.columns Pandas Data Structures >>> df.info() Info on DataFrame >>> df.iat([0],[0]) Number of non-NA values >>> df.count() Series 'Belgium' Summary A one-dimensional labeled array a 3 By Label Select single value by row & >>> df.sum() Sum of values capable of holding any data type b -5 >>> df.loc[[0], ['Country']] Cummulative sum of values 'Belgium' column labels >>> df.cumsum() >>> df.min()/df.max() Minimum/maximum values c 7 Minimum/Maximum index -

Pandas: Powerful Python Data Analysis Toolkit Release 0.25.3
pandas: powerful Python data analysis toolkit Release 0.25.3 Wes McKinney& PyData Development Team Nov 02, 2019 CONTENTS i ii pandas: powerful Python data analysis toolkit, Release 0.25.3 Date: Nov 02, 2019 Version: 0.25.3 Download documentation: PDF Version | Zipped HTML Useful links: Binary Installers | Source Repository | Issues & Ideas | Q&A Support | Mailing List pandas is an open source, BSD-licensed library providing high-performance, easy-to-use data structures and data analysis tools for the Python programming language. See the overview for more detail about whats in the library. CONTENTS 1 pandas: powerful Python data analysis toolkit, Release 0.25.3 2 CONTENTS CHAPTER ONE WHATS NEW IN 0.25.2 (OCTOBER 15, 2019) These are the changes in pandas 0.25.2. See release for a full changelog including other versions of pandas. Note: Pandas 0.25.2 adds compatibility for Python 3.8 (GH28147). 1.1 Bug fixes 1.1.1 Indexing • Fix regression in DataFrame.reindex() not following the limit argument (GH28631). • Fix regression in RangeIndex.get_indexer() for decreasing RangeIndex where target values may be improperly identified as missing/present (GH28678) 1.1.2 I/O • Fix regression in notebook display where <th> tags were missing for DataFrame.index values (GH28204). • Regression in to_csv() where writing a Series or DataFrame indexed by an IntervalIndex would incorrectly raise a TypeError (GH28210) • Fix to_csv() with ExtensionArray with list-like values (GH28840). 1.1.3 Groupby/resample/rolling • Bug incorrectly raising an IndexError when passing a list of quantiles to pandas.core.groupby. DataFrameGroupBy.quantile() (GH28113). -

Image Processing with Scikit-Image Emmanuelle Gouillart
Image processing with scikit-image Emmanuelle Gouillart Surface, Glass and Interfaces, CNRS/Saint-Gobain Paris-Saclay Center for Data Science Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features The world is getting more and more visual Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features The world is getting more and more visual Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features The world is getting more and more visual Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features The world is getting more and more visual Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features The world is getting more and more visual Principle Some features The world is getting more and more visual Image processing Manipulating images in order to retrieve new images or image characteristics (features, measurements, ...) Often combined with machine learning Principle Some features scikit-image http://scikit-image.org/ A module of the Scientific Python stack Language: Python Core modules: NumPy, SciPy, matplotlib Application modules: scikit-learn, scikit-image, pandas, ... A general-purpose image processing library open-source (BSD) not an application (ImageJ) less specialized than other libraries (e.g. OpenCV for computer vision) 1 Principle Principle Some features 1 First steps from skimage import data , io , filter image = data . -

Zero-Cost, Arrow-Enabled Data Interface for Apache Spark
Zero-Cost, Arrow-Enabled Data Interface for Apache Spark Sebastiaan Jayjeet Aaron Chu Ivo Jimenez Jeff LeFevre Alvarez Chakraborty UC Santa Cruz UC Santa Cruz UC Santa Cruz Rodriguez UC Santa Cruz [email protected] [email protected] [email protected] Leiden University [email protected] [email protected] Carlos Alexandru Uta Maltzahn Leiden University UC Santa Cruz [email protected] [email protected] ABSTRACT which is a very expensive operation; (2) data processing sys- Distributed data processing ecosystems are widespread and tems require new adapters or readers for each new data type their components are highly specialized, such that efficient to support and for each new system to integrate with. interoperability is urgent. Recently, Apache Arrow was cho- A common example where these two issues occur is the sen by the community to serve as a format mediator, provid- de-facto standard data processing engine, Apache Spark. In ing efficient in-memory data representation. Arrow enables Spark, the common data representation passed between op- efficient data movement between data processing and storage erators is row-based [5]. Connecting Spark to other systems engines, significantly improving interoperability and overall such as MongoDB [8], Azure SQL [7], Snowflake [11], or performance. In this work, we design a new zero-cost data in- data sources such as Parquet [30] or ORC [3], entails build- teroperability layer between Apache Spark and Arrow-based ing connectors and converting data. Although Spark was data sources through the Arrow Dataset API. Our novel data initially designed as a computation engine, this data adapter interface helps separate the computation (Spark) and data ecosystem was necessary to enable new types of workloads. -

Pandas Writing Data to Excell Spreadsheet
Pandas Writing Data To Excell Spreadsheet Nico automatizes rompingly while pandanaceous Orin elasticizing unheededly or flocculated cannily. Cold-hearted Dewitt Boyduntack sprawl her summersault some canticles? so vulnerably that Yehudi proclaim very distinctly. How auld is Shalom when caryatidal and slate When there are currently being written to a csv seems to increase or writing data to pandas to_excel function After you have data manipulation in excel spreadsheet, write to excel, i permanently delete. Python write two have done by writing them. As you can see my profile, i have good experience of python so i am sure i can make it soon Please chat with me and discuss Mehr. In python package is that writes to our mission: it is a text file is quite a single worksheet. This website uses cookies to improve your experience. My web application object that allows objects. Subscribe for writing large amounts of spreadsheet format across all rows whose sum. Text Import Wizard was more information about using the wizard. Python Data Analysis Library. In this case, mine was show a single Worksheet in the Workbook. Excel is a well known and really good user interface for many tasks. These types of spreadsheets can be used to perform calculations or create diagrams, for example. This out there are working with one reason why our files to python panda will try enabling it in. You have a spreadsheet with excel? Clustered indexes in this particular table, writing column number is. You much also use Python! This violin especially steep when can feel blocked on appropriate next step. -

Interaction Between SAS® and Python for Data Handling and Visualization Yohei Takanami, Takeda Pharmaceuticals
Paper 3260-2019 Interaction between SAS® and Python for Data Handling and Visualization Yohei Takanami, Takeda Pharmaceuticals ABSTRACT For drug development, SAS is the most powerful tool for analyzing data and producing tables, figures, and listings (TLF) that are incorporated into a statistical analysis report as a part of Clinical Study Report (CSR) in clinical trials. On the other hand, in recent years, programming tools such as Python and R have been growing up and are used in the data science industry, especially for academic research. For this reason, improvement in productivity and efficiency gain can be realized with the combination and interaction among these tools. In this paper, basic data handling and visualization techniques in clinical trials with SAS and Python, including pandas and SASPy modules that enable Python users to access SAS datasets and use other SAS functionalities, are introduced. INTRODUCTION SAS is fully validated software for data handling, visualization and analysis and it has been utilized for long periods of time as a de facto standard in drug development to report the statistical analysis results in clinical trials. Therefore, basically SAS is used for the formal analysis report to make an important decision. On the other hand, although Python is a free software, there are tremendous functionalities that can be utilized in broader areas. In addition, Python provides useful modules to enable users to access and handle SAS datasets and utilize SAS modules from Python via SASPy modules (Nakajima 2018). These functionalities are very useful for users to learn and utilize both the functionalities of SAS and Python to analyze the data more efficiently. -

Python Data Processing with Pandas
Python Data Processing with Pandas CSE 5542 Introduc:on to Data Visualizaon Pandas • A very powerful package of Python for manipulang tables • Built on top of numpy, so is efficient • Save you a lot of effort from wri:ng lower python code for manipulang, extrac:ng, and deriving tables related informaon • Easy visualizaon with Matplotlib • Main data structures – Series and DataFrame • First thing first • Series: an indexed 1D array • Explicit index • Access data • Can work as a dic:onary • Access and slice data DataFrame Object • Generalized two dimensional array with flexible row and column indices DataFrame Object • Generalized two dimensional array with flexible row and column indices DataFrame Object • From Pandas Series DataFrame Object • From Pandas Series DataFrame Object • Another example Viewing Data • View the first or last N rows Viewing Data • Display the index, columns, and data Viewing Data • Quick stas:cs (for columns A B C D in this case) Viewing Data • Sor:ng: sort by the index (i.e., reorder columns or rows), not by the data in the table column Viewing Data • Sor:ng: sort by the data values Selecng Data • Selec:ng using a label Selecng Data • Mul:-axis, by label Selecng Data • Mul:-axis, by label Slicing: last included Selecng Data • Select by posi:on Selecng Data • Boolean indexing Selecng Data • Boolean indexing Seng Data • Seng a new column aligned by indexes Seng Data Operaons • Descrip:ve stas:cs – Across axis 0 (rows), i.e., column mean – Across axis 1 (column), i.e., row mean Operaons • Apply • Histogram Merge Tables • Join Merge Tables • Append Grouping File I/O • CSV File I/O • Excel . -

Introduction to Python for Econometrics, Statistics and Data Analysis 4Th Edition
Introduction to Python for Econometrics, Statistics and Data Analysis 4th Edition Kevin Sheppard University of Oxford Thursday 31st December, 2020 2 - ©2020 Kevin Sheppard Solutions and Other Material Solutions Solutions for exercises and some extended examples are available on GitHub. https://github.com/bashtage/python-for-econometrics-statistics-data-analysis Introductory Course A self-paced introductory course is available on GitHub in the course/introduction folder. Solutions are avail- able in the solutions/introduction folder. https://github.com/bashtage/python-introduction/ Video Demonstrations The introductory course is accompanied by video demonstrations of each lesson on YouTube. https://www.youtube.com/playlist?list=PLVR_rJLcetzkqoeuhpIXmG9uQCtSoGBz1 Using Python for Financial Econometrics A self-paced course that shows how Python can be used in econometric analysis, with an emphasis on financial econometrics, is also available on GitHub in the course/autumn and course/winter folders. https://github.com/bashtage/python-introduction/ ii Changes Changes since the Fourth Edition • Added a discussion of context managers using the with statement. • Switched examples to prefer the context manager syntax to reflect best practices. iv Notes to the Fourth Edition Changes in the Fourth Edition • Python 3.8 is the recommended version. The notes require Python 3.6 or later, and all references to Python 2.7 have been removed. • Removed references to NumPy’s matrix class and clarified that it should not be used. • Verified that all code and examples work correctly against 2020 versions of modules. The notable pack- ages and their versions are: – Python 3.8 (Preferred version), 3.6 (Minimum version) – NumPy: 1.19.1 – SciPy: 1.5.3 – pandas: 1.1 – matplotlib: 3.3 • Expanded description of model classes and statistical tests in statsmodels that are most relevant for econo- metrics. -

Cheat Sheet: the Pandas Dataframe Object
Cheat Sheet: The pandas DataFrame Object Preliminaries Get your data into a DataFrame Always start by importing these Python modules Instantiate an empty DataFrame import numpy as np df = DataFrame() import matplotlib.pyplot as plt import pandas as pd Load a DataFrame from a CSV file from pandas import DataFrame, Series df = pd.read_csv('file.csv') # often works Note: these are the recommended import aliases df = pd.read_csv('file.csv', header=0, Note: you can put these into a PYTHONSTARTUP file index_col=0, quotechar='"', sep=':', na_values = ['na', '-', '.', '']) Note: refer to pandas docs for all arguments Cheat sheet conventions Get data from inline CSV text to a DataFrame from io import StringIO Code examples data = """, Animal, Cuteness, Desirable # Code examples are found in yellow boxes row-1, dog, 8.7, True row-2, cat, 9.5, True In the code examples, typically I use: row-3, bat, 2.6, False""" s to represent a pandas Series object; df = pd.read_csv(StringIO(data), header=0, df to represent a pandas DataFrame object; index_col=0, skipinitialspace=True) idx to represent a pandas Index object. Note: skipinitialspace=True allows for a pretty layout Also: t – tuple, l – list, b – Boolean, i – integer, a – numpy array, st – string, d – dictionary, etc. Load DataFrames from a Microsoft Excel file # Each Excel sheet in a Python dictionary workbook = pd.ExcelFile('file.xlsx') d = {} # start with an empty dictionary The conceptual model for sheet_name in workbook.sheet_names: df = workbook.parse(sheet_name) d[sheet_name] = df DataFrame object: is a two-dimensional table of data with column and row indexes (something like a spread Note: the parse() method takes many arguments like sheet). -

R Or Python? Dilemma, Dilemma
Paper SM01 R or Python? Dilemma, Dilemma Elsa Lozachmeur, Idorsia, Basel, Switzerland Nicolas Dupuis, Sanofi, Basel, Switzerland ABSTRACT RTM and PythonTM are the new kids in pharma town. These Open Source programming languages seem to be in every discussion lately. We see attempts in our industry to switch to them, replacing SAS®. They indeed come with advantages: licensing cost cut, a large community of users and developers, enabling their fast and impressive development. What are they good at in reality? What are their respective strengths? How do they compare to one another? And even more important, what are their current limitations? What can you do with SAS that you cannot do with R or Python, in your day-to-day Statistical Programmer life? Using our current personal understanding, we will take concrete examples with classic tasks and see how it can be done today with R and Python. INTRODUCTION As statistical programmers, we produce datasets (e.g. SDTM, ADaM), tables, listing and figures. In this paper, we will go through some classic tasks done in SAS and show you what it would look like if we had used R or Python instead. As in SAS, the code provided in this paper can be improved or other approaches could be used. R code has been tested in R Studio and Python code in Jupyter Notebook. WHAT’S R? Released in 1995, R is a popular, open-source language specialized in statistical analysis that can be extended by packages almost at will. R is commonly used with RStudio, a comfortable development environment that can be used locally or in a client-server installation via a web browser.