Utility of Trna Genes from the Complete Mitochondrial Genome of Psetta Maxima for Implying a Possible Sister-Group Relationship
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Updated Checklist of Marine Fishes (Chordata: Craniata) from Portugal and the Proposed Extension of the Portuguese Continental Shelf
European Journal of Taxonomy 73: 1-73 ISSN 2118-9773 http://dx.doi.org/10.5852/ejt.2014.73 www.europeanjournaloftaxonomy.eu 2014 · Carneiro M. et al. This work is licensed under a Creative Commons Attribution 3.0 License. Monograph urn:lsid:zoobank.org:pub:9A5F217D-8E7B-448A-9CAB-2CCC9CC6F857 Updated checklist of marine fishes (Chordata: Craniata) from Portugal and the proposed extension of the Portuguese continental shelf Miguel CARNEIRO1,5, Rogélia MARTINS2,6, Monica LANDI*,3,7 & Filipe O. COSTA4,8 1,2 DIV-RP (Modelling and Management Fishery Resources Division), Instituto Português do Mar e da Atmosfera, Av. Brasilia 1449-006 Lisboa, Portugal. E-mail: [email protected], [email protected] 3,4 CBMA (Centre of Molecular and Environmental Biology), Department of Biology, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal. E-mail: [email protected], [email protected] * corresponding author: [email protected] 5 urn:lsid:zoobank.org:author:90A98A50-327E-4648-9DCE-75709C7A2472 6 urn:lsid:zoobank.org:author:1EB6DE00-9E91-407C-B7C4-34F31F29FD88 7 urn:lsid:zoobank.org:author:6D3AC760-77F2-4CFA-B5C7-665CB07F4CEB 8 urn:lsid:zoobank.org:author:48E53CF3-71C8-403C-BECD-10B20B3C15B4 Abstract. The study of the Portuguese marine ichthyofauna has a long historical tradition, rooted back in the 18th Century. Here we present an annotated checklist of the marine fishes from Portuguese waters, including the area encompassed by the proposed extension of the Portuguese continental shelf and the Economic Exclusive Zone (EEZ). The list is based on historical literature records and taxon occurrence data obtained from natural history collections, together with new revisions and occurrences. -

Order ZEIFORMES PARAZENIDAE Parazens P.C
click for previous page Zeiformes: Parazenidae 1203 Order ZEIFORMES PARAZENIDAE Parazens P.C. Heemstra, South African Institute for Aquatic Biodiversity, South Africa iagnostic characters: Small to moderate-sized (to 30 cm) oblong fishes, the head and body com- Dpressed; body depth slightly less than head length, contained 2.6 to 2.9 times in standard length; head naked, the bones thin and soft; opercular bones weakly serrate; mouth large, terminal, the upper jaw extremely protrusile; maxilla widely expanded posteriorly, and mostly exposed when mouth is closed; no supramaxilla; jaws with 1 or 2 rows of small, slender, conical teeth; vomer with a few short stout teeth;gill rakers (including rudiments) 2 on upper limb, 8 on lower limb.Eye diameter about 1/3 head length and slightly less than snout length.Branchiostegal rays 7.Dorsal fin divided, with 8 slender spines and 26 to 30 soft rays; anal fin with 1 minute spine and 30 to 32 soft rays; dorsal-, anal-, and pectoral-fin rays un- branched; caudal fin forked, with 11 principal rays and 9 branched rays; pectoral fin with 15 or 16 rays, shorter than eye diameter; pelvic fins with 1 unbranched and 5 or 6 branched soft rays, but no spine, fin origin posterior to a vertical at pectoral-fin base. Scales moderate in size, weakly ctenoid, and deciduous; 2 lateral lines originating on body at upper end of operculum and running posteriorly about 4 scale rows apart, gradually converging to form a single line on caudal peduncle. Caudal peduncle stout, the least depth about equal to its length and slightly less than eye diameter.Vertebrae 34.Colour: body reddish or silvery; large black blotch on anterior margin of dorsal fin. -

Percomorph Phylogeny: a Survey of Acanthomorphs and a New Proposal
BULLETIN OF MARINE SCIENCE, 52(1): 554-626, 1993 PERCOMORPH PHYLOGENY: A SURVEY OF ACANTHOMORPHS AND A NEW PROPOSAL G. David Johnson and Colin Patterson ABSTRACT The interrelationships of acanthomorph fishes are reviewed. We recognize seven mono- phyletic terminal taxa among acanthomorphs: Lampridiformes, Polymixiiformes, Paracan- thopterygii, Stephanoberyciformes, Beryciformes, Zeiformes, and a new taxon named Smeg- mamorpha. The Percomorpha, as currently constituted, are polyphyletic, and the Perciformes are probably paraphyletic. The smegmamorphs comprise five subgroups: Synbranchiformes (Synbranchoidei and Mastacembeloidei), Mugilomorpha (Mugiloidei), Elassomatidae (Elas- soma), Gasterosteiformes, and Atherinomorpha. Monophyly of Lampridiformes is justified elsewhere; we have found no new characters to substantiate the monophyly of Polymixi- iformes (which is not in doubt) or Paracanthopterygii. Stephanoberyciformes uniquely share a modification of the extrascapular, and Beryciformes a modification of the anterior part of the supraorbital and infraorbital sensory canals, here named Jakubowski's organ. Our Zei- formes excludes the Caproidae, and characters are proposed to justify the monophyly of the group in that restricted sense. The Smegmamorpha are thought to be monophyletic principally because of the configuration of the first vertebra and its intermuscular bone. Within the Smegmamorpha, the Atherinomorpha and Mugilomorpha are shown to be monophyletic elsewhere. Our Gasterosteiformes includes the syngnathoids and the Pegasiformes -

Proceedings of the Indiana Academy of Science 1 1 8(2): 143—1 86
2009. Proceedings of the Indiana Academy of Science 1 1 8(2): 143—1 86 THE "LOST" JORDAN AND HAY FISH COLLECTION AT BUTLER UNIVERSITY Carter R. Gilbert: Florida Museum of Natural History, University of Florida, Gainesville, Florida 32611 USA ABSTRACT. A large fish collection, preserved in ethanol and assembled by Drs. David S. Jordan and Oliver P. Hay between 1875 and 1892, had been stored for over a century in the biology building at Butler University. The collection was of historical importance since it contained some of the earliest fish material ever recorded from the states of South Carolina, Georgia, Mississippi and Kansas, and also included types of many new species collected during the course of this work. In addition to material collected by Jordan and Hay, the collection also included specimens received by Butler University during the early 1880s from the Smithsonian Institution, in exchange for material (including many types) sent to that institution. Many ichthyologists had assumed that Jordan, upon his departure from Butler in 1879. had taken the collection. essentially intact, to Indiana University, where soon thereafter (in July 1883) it was destroyed by fire. The present study confirms that most of the collection was probably transferred to Indiana, but that significant parts of it remained at Butler. The most important results of this study are: a) analysis of the size and content of the existing Butler fish collection; b) discovery of four specimens of Micropterus coosae in the Saluda River collection, since the species had long been thought to have been introduced into that river; and c) the conclusion that none of Jordan's 1878 southeastern collections apparently remain and were probably taken intact to Indiana University, where they were lost in the 1883 fire. -

2019-JMIH-Program-Book-MASTER
W:\CNCP\People\Richardson\FY19\JMIH - Rochester NY\Program\2018 JMIH Program Book.pub 2 Organizing Societies American Elasmobranch Society 34th Annual Meeting President: Dave Ebert Treasurer: Christine Bedore Secretary: Tonya Wiley Editor and Webmaster: Chuck Bangley Immediate Past President: Dean Grubbs American Society of Ichthyologists and Herpetologists 98th Annual Meeting President: Kathleen Cole President Elect: Chris Beachy Past President: Brian Crother Prior Past President: Carole Baldwin Treasurer: Katherine Maslenikov Secretary: Prosanta Chakrabarty Editor: W. Leo Smith Herpetologists’ League 76th Annual Meeting President: Willem Roosenburg Vice-President: Susan Walls Immediate Past President: David Sever (deceased) Secretary: Renata Platenburg Treasurer: Laurie Mauger Communications Secretary: Max Lambert Herpetologica Editor: Stephen Mullin Herpetological Monographs Editor: Michael Harvey Society for the Study of Amphibians and Reptiles 61th Annual Meeting President: Marty Crump President-Elect: Kirsten Nicholson Immediate Past-President: Richard Shine Secretary: Marion R. Preest Treasurer: Ann V. Paterson Publications Secretary: Cari-Ann Hickerson 3 Thanks to our Sponsors! PARTNER SPONSOR SUPPORTER SPONSOR 4 We would like to thank the following: Local Hosts Alan Savitzky, Utah State University, LHC Co-Chair Catherine Malone, Utah State University, LHC Co-Chair Diana Marques, Local Host Logo Artist Marty Crump, Utah State University Volunteers We wish to thank the following volunteers who have helped make the Joint Meeting -

Order Myctophiformes, Lanternfishes
Order Myctophiformes, lanternfishes • 241 species, 35 genera, 2 families • Deep sea pelagic and benthic, numerically dominant in deep sea habitats • Large terminal mouth (reminiscent of anchovy) • Adipose fin present • Compressed head and body (Myctophiformes = nose serpent shape) Lampridiformes • Large eyes Percopsiformes • Photophores Acanthomorpha •Hollow unsegmented spines on dorsal and anal fins •Rostal and premaxilla cartilidge and ligaments allow greater jaw protrusability Order Lampridiformes, opahs and oarfish Order Lampridiformes, opahs and oarfish • Oarfish • 19 species, 12 genera, 7 families – Longest teleost – over 30 feet • no true spines in fins – Only one individual observed • unique upper jaw protrusion – alive, used amiiform maxilla not directly attached to swimming ethmoid or palentine • deep bodied or ribbon-like • pelagic and deep water marine 1 Order Percopsiformes, trout perch, pirate perch, cavefish • 3 families, 7 genera, 9 species • All freshwater • Few with adipose fins – one of the most derived fishes with them • Pirate perch (Aphredoderidae) – One species – Fairly extensive parental care – Anus migration • Cavefish (Amblyopsidae) Zeiformes – Reduction or loss of eyes Gadiformes – Sensory papillae on head, body and tail Acanthomorpha •Hollow unsegmented spines on – Anus migration dorsal and anal fins •Rostal and premaxilla cartilidge – Convergent evolution of cave fish and ligaments allow greater jaw and other cave characins, protrusability catfishes etc. Order Zeiformes Order Gadiiformes • Dories • 555 species, -
Field Identification Guide to the Living Marine Resources in Kenya
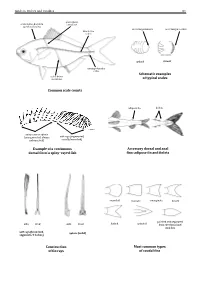
Guide to Orders and Families 81 lateral line scales above scales before dorsal fin outer margin smooth outer margin toothed (predorsal scales) lateral–line 114 scales cycloid ctenoidِّ scales circumpeduncular Schematic examples lateral line of typical scales scales below Common scale counts adipose fin finlets soft rays (segmented, spinyunbranched) rays or spines usually branched) (unsegmented, always Example of a continuous Accessory dorsal and anal dorsal fin of a spiny–rayed fish fins: adipose fin and finlets rounded truncate emarginate lunate side front side front from the dorsal and pointed and separated forked pointed soft rays (branched, spines (solid) segments, 2 halves) anal fins Construction Most common types of fin rays of caudal fins 82 Bony Fishes GUIDE TO ORDERS AND FAMILIES Order ELOPIFORMES – Tarpons and allies Fin spines absent; a single dorsal fin located above middle of body; pelvic fins in abdominal position; lateral line present; 23–25 branchiostegal rays; upper jaw extending past eye; tip of snout not overhanging mouth; colour silvery. ELOPIDAE Page 121 very small scales Ladyfishes To 90 cm. Coastal marine waters and estuaries; pelagic. A single species included in the Guide to Species.underside of head large mouth gular plate MEGALOPIDAE Page 121 last ray long Tarpons large scales To 55 cm. Coastal marine waters and estuaries; pelagic. A single species included in the Guide to Species.underside of head gular plate Order ALBULIFORMES – Bonefishes Fin spines absent; a single dorsal fin located above middle of body; pelvic fins in abdominal position; lateral line present; 6–16 branchiostegal rays; upper jaw not extending as far as front of eye; tip of snout overhanging mouth; colour silvery. -

Digeneans (Trematoda) Parasitic in Freshwater Fishes (Osteichthyes) of the Lake Biwa Basin in Shiga Prefecture, Central Honshu, Japan
Digeneans (Trematoda) Parasitic in Freshwater Fishes (Osteichthyes) of the Lake Biwa Basin in Shiga Prefecture, Central Honshu, Japan Takeshi Shimazu1, Misako Urabe2 and Mark J. Grygier3 1 Nagano Prefectural College, 8–49–7 Miwa, Nagano City, Nagano 380–8525, Japan and 10486–2 Hotaka-Ariake, Azumino City, Nagano 399–8301, Japan E-mail: [email protected] 2 Department of Ecosystem Studies, School of Environmental Science, The University of Shiga Prefecture, 2500 Hassaka, Hikone City, Shiga 522–8533, Japan 3 Lake Biwa Museum, 1091 Oroshimo, Kusatsu City, Shiga 525–0001, Japan Abstract: The fauna of adult digeneans (Trematoda) parasitic in freshwater fishes (Osteichthyes) from the Lake Biwa basin in Shiga Prefecture, central Honshu, Japan, is studied from the literature and existing specimens. Twenty-four previously known, 2 new, and 4 unidentified species in 17 gen- era and 12 families are recorded. Three dubious literature records are also mentioned. All 30 con- firmed species, except Sanguinicolidae gen. sp. (Aporocotylidae), are described and figured. Life cy- cles are discussed where known. Philopinna kawamutsu sp. nov. (Didymozoidae) was found in the connective tissue between the vertebrae and the air bladder near the esophagus of Nipponocypris tem- minckii (Temminck and Schlegel) (Cyprinidae). Genarchopsis yaritanago sp. nov. (Derogenidae) was found in the intestine of Tanakia lanceolata (Temminck and Schlegel) (Cyprinidae). Asymphylodora innominata (Faust, 1924) comb. nov. is proposed for A. macrostoma Ozaki, 1925 (Lissorchiidae). A key to the families, genera, and species of these digeneans is provided. Host-parasite and parasite- host lists are given. Key words: adult digeneans, Trematoda, parasites, morphology, life cycle, Philopinna kawamutsu sp. -
Parasitic Copepods (Crustacea, Hexanauplia) on Fishes from the Lagoon Flats of Palmyra Atoll, Central Pacific
A peer-reviewed open-access journal ZooKeys 833: 85–106Parasitic (2019) copepods on fishes from the lagoon flats of Palmyra Atoll, Central Pacific 85 doi: 10.3897/zookeys.833.30835 RESEARCH ARTICLE http://zookeys.pensoft.net Launched to accelerate biodiversity research Parasitic copepods (Crustacea, Hexanauplia) on fishes from the lagoon flats of Palmyra Atoll, Central Pacific Lilia C. Soler-Jiménez1, F. Neptalí Morales-Serna2, Ma. Leopoldina Aguirre- Macedo1,3, John P. McLaughlin3, Alejandra G. Jaramillo3, Jenny C. Shaw3, Anna K. James3, Ryan F. Hechinger3,4, Armand M. Kuris3, Kevin D. Lafferty3,5, Victor M. Vidal-Martínez1,3 1 Laboratorio de Parasitología, Centro de Investigación y de Estudios Avanzados del IPN (CINVESTAV- IPN) Unidad Mérida, Carretera Antigua a Progreso Km. 6, Mérida, Yucatán C.P. 97310, México 2 CONACYT, Centro de Investigación en Alimentación y Desarrollo, Unidad Académica Mazatlán en Acuicultura y Manejo Ambiental, Av. Sábalo Cerritos S/N, Mazatlán 82112, Sinaloa, México 3 Department of Ecology, Evolution and Marine Biology and Marine Science Institute, University of California, Santa Barbara CA 93106, USA 4 Scripps Institution of Oceanography-Marine Biology Research Division, University of California, San Diego, La Jolla, California 92093 USA 5 Western Ecological Research Center, U.S. Geological Survey, Marine Science Institute, University of California, Santa Barbara CA 93106, USA Corresponding author: Victor M. Vidal-Martínez ([email protected]) Academic editor: Danielle Defaye | Received 25 October 2018 | -

Blenniidae; Teleostei) ⁎ Shang-Yin Vanson Liua, , Bruno Frédérichb,1, Sébastien Lavouéc, Jonathan Changd, Mark V
Molecular Phylogenetics and Evolution 125 (2018) 138–146 Contents lists available at ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev Buccal venom gland associates with increased of diversification rate in the T fang blenny fish Meiacanthus (Blenniidae; Teleostei) ⁎ Shang-Yin Vanson Liua, , Bruno Frédérichb,1, Sébastien Lavouéc, Jonathan Changd, Mark V. Erdmanne, Gusti Ngurah Mahardikaf, Paul H. Barberd a Department of Marine Biotechnology and Resources, National Sun Yat-Sen University, Kaohsiung 80424, Taiwan b Laboratoire d’Océanologie, UR FOCUS, Université de Liège, 4000 Liège, Belgium c Institute of Oceanography, National Taiwan University, Roosevelt Road, Taipei 10617, Taiwan d Department of Ecology and Evolutionary Biology, University of California Los Angeles, Los Angeles, CA 90095-7239, USA e Conservation International Indonesia Marine Program, 80235 Bali, Indonesia f The Indonesian Biodiversity Research Centre, The Animal Biomedical and Molecular Biology Laboratory of Udayana University, Jl Sesetan-Markisa 6, Denpasar, Bali, Indonesia ARTICLE INFO ABSTRACT Keywords: At the macroevolutionary level, many mechanisms have been proposed to explain explosive species diversifi- Macroevolution cation. Among them morphological and/or physiological novelty is considered to have a great impact on the Innovation tempo and the mode of diversification. Meiacanthus is a genus of Blenniidae possessing a unique buccal venom fi Diversi cation rate shift gland at the base of an elongated canine tooth. This unusual trait has been hypothesized to aid escape from Coral reef fishes predation and thus potentially play an important role in their pattern of diversification. Here, we produce the first time-calibrated phylogeny of Blenniidae and we test the impact of two morphological novelties on their diversification, i.e. -

1 Title 1 Spring-Fed Streams As Flow Refugia: Changes in Fish
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.26.062968; this version posted April 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title 2 Spring-fed streams as flow refugia: Changes in fish assemblages after a rainfall event 3 4 Authors 5 Masaru Sakai1 | Ryoshiro Wakiya2 | Gosuke Hoshi3 6 7 Affiliations 8 1 National Institute for Environmental Studies, Japan (Fukushima Branch), Fukushima, 9 Japan 10 2 Atmosphere and Ocean Research Institute, the University of Tokyo, Chiba, Japan 11 3 Graduate School of Science and Engineering, Chuo University, Tokyo, Japan 12 13 Correspondence 14 Masaru Sakai, National Institute for Environmental Studies, Japan (Fukushima Branch), 15 10-2 Fukasaku, Miharu, Tamura District, Fukushima 963-7700, Japan 16 Email: [email protected] 17 ORCID iD: 0000-0001-5361-0978 18 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.04.26.062968; this version posted April 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 19 Abstract 20 Understanding the structure and function of flow refugia in river networks is critical to 21 freshwater conservation in the context of anthropogenic changes in flow regimes. Safety 22 concerns associated with field data collection during floods have largely hindered 23 advances in assessing flow refugia for fishes; however, spring-fed streams can be safely 24 surveyed during rainfall events owing to their stable flow regimes. -

2014 Annual Meeting Program Oral Presentation
2014 Annual Meeting Program Papers to be presented at the 47th Annual Meeting of the Ichthyological Society of Japan at Kanagawa Prefectural Museum of Natural History, Novenber 14-17, 2014 Oral Presentation (Each presentation has been allowed 15 min, i.e. 12 min for presentation and 3 min for questions; *speaker) Oral Presentation Room #1 (SEISA Museum Theater, 1st Floor) November 15 (Saturday) Ecology, Conservation 1 09:30- Male and bisexual life history polymorphisms in anadromous fishes-II: Why does Arctic lamprey adopt a bisexual polymorphism? Akira GOTO* and Yuji YAMAZAKI 2 09:45- Habitat use of alien piscivorous chub establishing in small agricultural ditches Yoshihisa KURITA*, Norio ONIKURA and Ryutei INUI 3 10:00- Reproductive characteristics of Pseudorasbora sp. ,"Ushi-motsugo" under captive condition Moe MIYANISHI*, Tetsuro KITAGAWA, Yuka ODA and Kazumi HOSOYA 4 10:15- Spawning habitat selectivity of Honmoroko in inlets to Nishinoko and Ibanaiko Lagoon Takeshi KIKKO*, Daisuke ISHIZAKI, Yoshitaka KATAOKA and Yoshiaki KAI 5 10:30- Ecology of Lefua echigonia in the headwaters of the Tatarazawa, Atugi City-2 Hidetaka SUMIKURA* and Naoyuki SUGURO 6 10:45- Growth and longevity of the Japanese eight-barbel loach in a small marsh in the Kako River system Shigeru AOYAMA*, Tomohiro TABATA, Toshio DOI and Jinsuke AKADA <Break> <General Meeting of Members, 11:15-12:00 (Room #1)> <Lecture of the Awardee, 12:00-12:30 (Room #1)> <Lunch> <Poster Session Core Time on Odd-Numbered Paper, 13:30-14:30> Ecology, Conservation 7 14:30- Life history