Discovering Hidden Traps
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Reporter272web.Pdf
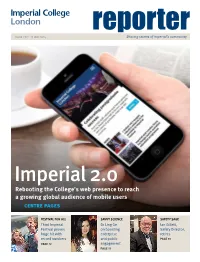
Issue 272 ▸ 15 may 2014 reporterSharing stories of Imperial’s community Imperial 2.0 Rebooting the College’s web presence to reach a growing global audience of mobile users → centre pages FESTIVAL FOR ALL SAVVY SCIENCE SAFETY SAGE Third Imperial Dr Ling Ge Ian Gillett, Festival proves on boosting Safety Director, huge hit with enterprise retires record numbers and public PAGE 10 PAGE 12 engagement PAGE 11 2 >> newsupdate www.imperial.ac.uk/reporter | reporter | 15 May 2014 • issue 272 Renewed drive for engineering through inspirational role models; and improve and increase Imperial’s recognition for equality in UK science promoting gender equality through Athena SWAN Charter awards. Imperial has joined a campaign led by the Chancellor Professor Debra Humphris, Vice Provost (Education) EDITOR’S CORNER to boost participation in technology and engineering at Imperial, said: “We want to help shatter myths and careers among women. change perceptions about women in STEM. It’s fantastic to get the Chancellor’s backing for these goals. Digital The ‘Your Life’ initiative brings together government, business, professional bodies and leading educational wonder institutions who are all working to improve We want to help shatter myths opportunities for women in science, technology, and change perceptions.” Do you remember those engineering and maths (STEM). The scheme was tentative steps when launched by Chancellor George Osborne at the Science you first dipped your Museum on 7 May. “Meeting this challenge will not be easy. It will toes into the World Wide As part of the campaign Imperial has pledged to: require a concerted effort throughout the College. -

Modelling and Optimising Gaas/Al (X) Ga (1-X) As Multiple Quantum Well
University of London Imperial College of Science, Technology and Medicine Department of Physics Modelling and Optimising GaAs/AlxGa1 xAs − Multiple Quantum Well Solar Cells James P. Connolly arXiv:1006.1053v1 [cond-mat.mes-hall] 5 Jun 2010 Submitted in part fulfilment of the requirements for the degree of Doctor of Philosophy in Science of the University of London and the Diploma of Imperial College, January 1997 2 Abstract The quantum well solar cell (QWSC) is a p - i - n solar cell with quantum wells in the intrinsic region. Previous work has shown that QWSCs have a greater open circuit voltage (Voc) than would be provided by a cell with the quantum well effective bandgap. This suggests that the fundamental efficiency limits of QWSCs are greater than those of single bandgap solar cells. The following work investigates QWSCs in the GaAs/AlxGa1−xAs materials system. The design and optimisation of a QWSC in this system requires studies of the voltage and current dependencies on the aluminium fraction. QWSCs with different aluminium fractions have been studied and show an increasing Voc with increasing barrier aluminium composition. The QE however decreases with increasing aluminium composition. We de- velop a model of the QE to test novel QWSC designs with a view to minimising this problem. This work concentrates on two design changes. The first deals with com- positionally graded structures in which the bandgap varies with position. This bandgap variation introduces an quasi electric field which can be used to increase minority carrier collection in the low efficiency p and n layers. This technique also increases the light flux reaching the highly efficient depletion regions. -

Didier Queloz Comes to Cambridge
CavMag JANUARY 2013 Issue 9 News from the Cavendish Laboratory Inside... Editorial 2 Ground-breaking for the Battcock Centre for Experimental 3 Astrophysics DNA Origami Nanopores 4 The Higgs Boson and a Challenge 5 for Supersymmetry Spin Ices and Magnetic 6 Monopoles Wedge Issue 7 50th Anniversary of Brian 8 Josephson’s Nobel Prize Discovery Eryl Wynn-Williams and the Scale- 9 of-Two Counter First Winton Symposium - Energy 10 Efficiency Athene Donald Blogs Physics at 11 Work The Sutton Trust Summer Schools 12 in Physics Outreach and Educational Events 13 Development: Cavendish 3 - 14 Principles for Design Cavendish News 15 Didier Queloz comes to Cambridge xoplanet research is a his appointment is of great themselves at the forefront of this relatively new discipline. significance for future astronomical remarkable quest by capitalising on It started in 1995 with the research in Cambridge. As he synergies and potentials present at first definitive detection writes: Cambridge and in the country. Eof a planet orbiting a normal star beyond the Solar System ‘The search for planetary systems For the next decade, my main by Michel Mayor and Didier orbiting other stars and particularly research objective is to conduct Queloz. Since then the field has the quest to find planets similar a coherent effort towards the expanded exponentially into a to the Earth is one of the great detection and characterisation of major world-wide activity - it scientific, technological and planets with the goal of advancing is one of the areas of modern philosophical undertakings of our our understanding of their Main image: An astrophysics that has particularly time. -

The Efficiency of Photovoltaic Systems
UNIVERSITY OF SOUTHAMPTON The Efficiency of Photovoltaic Systems by Athanasios Gousiopoulos A thesis for the degree of Doctor of Philosophy in Electrical and Electronic Engineering Faculty of Physical Sciences and Engineering Department of Electronics and Computer Science November 2016 UNIVERSITY OF SOUTHAMPTON ABSTRACT FACULTY OF PHYSICAL SCIENCES AND ENGINEERING DEPARTMENT OF ELECTRONICS & COMPUTER SCIENCE Doctor of Philosophy The Efficiency of Photovoltaic Systems by Athanasios Gousiopoulos At this work the principle aim is to create new low cost hardware test circuitry that can emulate the behavior of solar cells under different insolation and temperature conditions. Another goal is to propose a new MPPT algorithm that could accurate and reliable estimate the maximum power operating point. The transcendental equation describing a solar cell IV characteristic contains current on both sides in a function of the form I = f (V, I). In this work, a current- independent voltage expression is derived for the maximum power point as a function of a new variable which is mathematically well defined. Validation is performed on four different photovoltaic modules. The best case scenario has shown a divergence in Pmpp of 0.08% while the worst 0.84%. The new method is examined for sensitivity, up to ± 5% on values of five fitting parameters (Rs, Rsh, n, Io, Iph), with Iph to have the higher impact effect (up to 5% error). Two topologies, of VBE multiplier, have been proposed that can reliably emulate solar cell operation. Analysis shows that both circuits have the potential to deliver good quality characteristic IV curves with small RMSE (10-3). The second improved VBE multiplier has the ability to operate over wider range of illumination and temperature conditions. -

Device Performance of Emerging Photovoltaic Materials (Version 1)
Device Performance of Emerging Photovoltaic Materials (Version 1) Item Type Article Authors Almora, Osbel; Baran, Derya; Bazan, Guillermo C.; Berger, Christian; Cabrera, Carlos I.; Catchpole, Kylie R.; Erten-Ela, Sule; Guo, Fei; Hauch, Jens; Ho-Baillie, Anita; Jacobsson, T. Jesper; Janssen, Rene A. J.; Kirchartz, Thomas; Kopidakis, Nikos; Li, Yongfang; Loi, Maria A.; Lunt, Richard R.; Mathew, Xavier; McGehee, Michael D.; Min, Jie; Mitzi, David B.; Nazeeruddin, Mohammad K.; Nelson, Jenny; Nogueira, Ana F.; Paetzold, Ulrich W.; Park, Nam-Gyu; Rand, Barry P.; Rau, Uwe; Snaith, Henry J.; Unger, Eva; Vaillant-Roca, Lídice; Yip, Hin-Lap; Brabec, Christoph J. Citation Almora, O., Baran, D., Bazan, G. C., Berger, C., Cabrera, C. I., Catchpole, K. R., … Brabec, C. J. (2020). Device Performance of Emerging Photovoltaic Materials (Version 1). Advanced Energy Materials, 2002774. doi:10.1002/aenm.202002774 Eprint version Publisher's Version/PDF DOI 10.1002/aenm.202002774 Publisher Wiley Journal Advanced Energy Materials Rights This is an open access article under the terms of the Creative Commons Attribution-NonCommercial License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes. Download date 01/10/2021 23:40:33 Item License http://creativecommons.org/licenses/by-nc/4.0/ Link to Item http://hdl.handle.net/10754/666279 PROGRESS REPORT www.advenergymat.de Device Performance of Emerging Photovoltaic Materials (Version 1) Osbel Almora,* Derya Baran, Guillermo C. Bazan, Christian Berger, Carlos I. Cabrera, Kylie R. Catchpole, Sule Erten-Ela, Fei Guo, Jens Hauch, Anita W. Y. Ho-Baillie, T. -

Solar Power for CO2 Mitigation Jenny Nelson, Ajay Gambhir and Ned Ekins-Daukes
Grantham institute for climate change Briefing paper no 11 January 2014 Solar power for CO2 mitigation JEnny nElson, AJAy GAmbhir And nEd Ekins-DaukEs Executive summary Solar power repreSentS a vaSt reSource which could, in principle, meet the world’s needs for low-carbon power generation many times over. the technology to generate solar power by conversion of light to electricity (pv) and conversion of light to power via heat (solar thermal) is already proven and widely deployed. the cost reductions in solar pv over the last ten years now make it competitive with conventional, fossil fuel based grid power in some locations, and it will soon be competitive in others, including the uK. Solar power is particularly relevant to the developing world where solar resource is high and solar power with storage is likely soon to become a more cost- effective option than diesel generators. recent growth in the use of photovoltaic (pv) technology (of around 40% per year) and rapid reduction in its cost (20% per doubling of capacity) has demonstrated the potential of solar power to deliver on a large scale. the Contents international energy agency (IEA) projects that solar power could generate executive summary ..................... 1 22% of the world’s electricity by 2050. this would remove a significant fraction of the growing global carbon dioxide (CO ) emissions from fossil generation. 2 introduction ............................. 2 Such a target is ambitious but achievable: the rate of growth of installed capacity needed to meet this target is much lower than the actual average Solar energy technology and growth rate over the last 20 years. -

Transfer State and Device Performance of Hybrid and Organic
Spectroscopic Studies of the Charge- Transfer State and Device Performance of Hybrid and Organic Solar Cells Flurin David Eisner A thesis submitted in partial fulfilment of the degree of Doctor of Philosophy Imperial College London Department of Physics April 2019 DECLARATION OF ORIGINALITY This thesis describes the work carried out between October 2015 and December 2018 in the Experimental Solid State Physics group of Imperial College London, under the supervision of Prof. Thomas D. Anthopoulos and Prof. Jenny Nelson. The material presented herein is the product of my own work, unless stated. Where information has been derived from other sources, this has been indicated in the thesis. COPYRIGHT DECLATION The copyright of this thesis rests with the author and is made available under a Creative Commons Attribution Non-Commercial No Derivatives License (CC BY-NC-ND). Researchers are free to copy, distribute or transmit the thesis on the condition that they attribute it, that they do not use it for commercial purposes and that they do not alter, transform or build upon it. For any reuse or redistribution, researchers must make clear to others the license terms of this work. Flurin Eisner London, April 2019 2 Abstract The last few years have witnessed a remarkable explosion in advances in the field of photovoltaics (PV) based on organic semiconductors as the light absorbing material, both in terms of device efficiency and in terms of the physical understanding of the underlying processes. This thesis aims to contribute to this field by examining two different types of solar cells based on light-absorbing organic semiconductors. -

Acknowledgment of Reviewers, 2019
Acknowledgment of Reviewers, 2019 PNAS would like to thank all the individuals who dedicated their considerable time and expertise to the journal by serving as reviewers in 2019. Their generous contribution is deeply appreciated. A Dean C. Adams Bahareh Ajami Karen Alim Jorge Alvarez-Solas Mu A Michael W. W. Adams Michael Akam Dan Alistarh James C. Alwine Rolf Aalto Michael Adams Masaki Akaogi Kari Alitalo Teresa Amabile Duur K. Aanen Philip Adams Schahram Akbarian Jennifer M. Alix-Garcia Rudolf Amann Maria Abascal Russell Adams Erol Akçay Saed Alizamir Katrin Amann-Winkel Adam R. Abate Mokhtar Adda-Bedia Seun Akeju Richard Alkire Susan G. Amara Cory Abate-Shen Louigi Addario-Berry Mark Akeson Eric Allan Luis A. Nunes Amaral Abul K. Abbas Donna Rose Addis Huda Akil David Alland Gaya K. Amarasinghe Jon Abbatt Zach N. Adelman Modupe Akinola Hunt Allcott Kapil Amarnath Patrick Abbot Hillel Adesnik Masashi Akiyama Benjamin L. Allen Richard Ambinder Karen C. Abbott William Neil Adger Takahiko Akiyama David T. Allen Sandro Ambuehl Larry Abbott Achyuta Adhvaryu Michael Aklin Eric E. Allen Ken-ichi Amemori Nicholas L. Abbott Claire L. Adida Georgiy Akopov Jenny Allen Jan Amend Zakia Abdelhamed Jess F. Adkins Klaus Aktories Karen N. Allen Seth A. Ament Mohamed H. Frederick R. Adler Alessandro Alabastri Micah Allen Stefano Amente Abdel-Rahman Nancy E. Adler Petri Ala-Laurila Nicola J. Allen Manuel R. Amieva Omar Abdel-Wahab Ralph Adolphs Richard Alba Paul M. Allen Sebastian Amigorena Ikuro Abe Tobias Adrian Joseph Albanesi Stefano Allesina Ariel Amir Guy J. Abel Markus Aebi Umberto Albarella Heidi Alleway Ido Amit Laurent Abel Shuchin Aeron Jawdat Al-Bassam David B. -
Open Letter - Gas Attack in Taxonomy
Open Letter - Gas Attack in Taxonomy Dear President von der Leyen, Vice-President Timmermans and Dombrovskis, Commissioner McGuinness, We are alarmed by a new proposal allowing fossil fuels into the EU taxonomy. This new proposal drafted by the European Commission suggests that fossil gas does no harm to the environment, and therefore could be included as Sustainable for Finance under the EU Taxonomy. These are baseless claims and are opposed to climate science. Introducing fossil gas in the EU taxonomy is firmly against the recommendations of the Commission’s Technical Expert Group and caves in to the demands of the gas lobby. The EU taxonomy was conceived as a science-based gold standard to avoid greenwashing. With such a proposal the taxonomy itself would become a greenwashing tool. Energy security, just transition, and grid reliability are critical for Europe, but are also fully addressed by other EU policies, funds, and regulations. The purpose of the EU Taxonomy is to correctly label green finance: this means following the best scientific evidence on an activity’s environmental impact. Counting gas as green ignores the significant environmental effects of methane, whose impact on climate change is up to 84 times greater than CO2 in a 20-year timeframe. This means if only 3% of the gas leaks, it can cause more warming than coal.1 Furthermore, many European gas companies do not properly measure methane emissions in their supply chain and are not seizing the available opportunities to reduce these emissions.2 We have no time for false solutions. This is why neither coal-to-gas nor cogeneration (CHP) should be included in the Taxonomy. -

Trustees' Report and Financial Statements 2014-15
TRUSTEES’ REPORT AND FINANCIAL STATEMENTS 1 Trustees’ report and financial statements For the year ended 31 March 2015 2 TRUSTEES’ REPORT AND FINANCIAL STATEMENTS Trustees Executive Director The Trustees of the Society are the Dr Julie Maxton members of its Council, who are elected Statutory Auditor by and from the Fellowship. Council is Deloitte LLP chaired by the President of the Society. Abbots House During 2014/15, the members of Council Abbey Street were as follows: Reading President RG1 3BD Sir Paul Nurse Bankers Treasurer The Royal Bank of Scotland Professor Anthony Cheetham 1 Princess Street London Physical Secretary EC2R 8BP Sir John Pethica* Professor Alexander Halliday** Investment Managers Rathbone Brothers PLC Biological Secretary 1 Curzon Street Sir John Skehel London Foreign Secretary W1J 5FB Professor Martyn Poliakoff CBE Internal Auditors Members of Council PricewaterhouseCoopers LLP Sir John Beddington CMG Cornwall Court Professor Geoffrey Boulton* 19 Cornwall Street Professor Andrea Brand Birmingham Professor Michael Cates B3 2DT Dame Athene Donald DBE Professor Carlos Frenk Professor Uta Frith DBE** Professor Joanna Haigh** Registered Charity Number 207043 Dame Wendy Hall DBE Registered address Dr Hermann Hauser** 6 – 9 Carlton House Terrace Dame Frances Kirwan DBE London SW1Y 5AG Professor Ottoline Leyser CBE Professor Angela McLean royalsociety.org Professor Georgina Mace CBE Professor Roger Owen Professor Timothy Pedley* Dame Nancy Rothwell DBE Professor Stephen Sparks CBE** Professor Ian Stewart** Dame Janet Thornton DBE** Professor John Wood* * Until 1 December 2014 ** From 1 December 2014 TRUSTEES’ REPORT AND FINANCIAL STATEMENTS 3 Contents President’s foreword ................................................ 4 Executive Director’s report ............................................ 5 Trustees’ report ................................................... 6 Promoting science and its benefits ................................... -

Next Generation Materials for Solar Photovoltaics
PROGRAMME NEXT GENERATION MATERIALS FOR SOLAR PHOTOVOLTAICS A 1-day Symposium Hosted by The Royal Society of Chemistry Energy Sector The Chemistry Centre, Burlington House, London 15 January 2020 This meeting covers recent advances in Solar Photovoltaics with a focus on materials for organic, inorganic and hybrid thin film cells. The day will comprise presentations by six guest speakers and seven contributing speakers and poster presentations during the lunch and coffee breaks. ORGANISING COMMITTEE Prof Trystan Watson Swansea University Dr Petra Cameron University of Bath Prof Martin Heeney Imperial College London Dr Ross Hatton University of Warwick 1 SPONSORS This meeting has been underwritten by the RSC Energy Sector interest group. Sponsors of student discount Our thanks to RSC-ESED for financial support towards the cost of overseas speakers Our thanks to SuperSolar for financing 10 student free tickets Feedback please to [email protected] 2 GUEST SPEAKERS Dr Eva Unger From lead-halide solution complexes to perovskite solar cell modules Eva Unger is the young investigator group leader for hybrid materials formation and scaling at Helmholtz Zentrum Berlin. Originally from Germany, Eva undertook her PhD at Uppsala University, Sweden and carried out postdoctoral work at Stanford University and Lund University through a stipend from the Swedish Marcus and Amalia Wallenberg Foundation. One of the goals of her group is to develop and optimize scalable deposition methods for hybrid perovskite semiconductors. Dr Theodoros Dimopoulos Emerging PV technologies based on abundant and low-environmental-impact materials Theodoros Dimopoulos is Senior Scientist at the Austrian Institute of Technology. His research involves solution and vacuum-based processing of transparent electrodes and solar absorbers, emerging photovoltaic materials and processes, in particular developing new materials, manufacturing processes and characterisation methods for thin-film solar cells. -

Membership of Sectional Committees 2015
Membership of Sectional Committees 2015 The main responsibility of the Sectional Committees is to select a short list of candidates for consideration by Council for election to the Fellowship. The Committees meet twice a year, in January and March. SECTIONAL COMMITTEE 1 [1963] SECTIONAL COMMITTEE 3 [1963] Mathematics Chemistry Chair: Professor Keith Ball Chair: Professor Anthony Stace Members: Members: Professor Philip Candelas Professor Varinder Aggarwal Professor Ben Green Professor Harry Anderson Professor John Hinch Professor Steven Armes Professor Christopher Hull Professor Paul Attfield Professor Richard Kerswell Professor Shankar Balasubramanian Professor Chandrashekhar Khare Professor Philip Bartlett Professor Steffen Lauritzen Professor Geoffrey Cloke Professor David MacKay Professor Peter Edwards Professor Robert MacKay Professor Malcolm Levitt Professor James McKernan Professor John Maier Professor Michael Paterson Professor Stephen Mann Professor Mary Rees Professor David Manolopoulos Professor John Toland Professor Paul O’Brien Professor Srinivasa Varadhan Professor David Parker Professor Alex Wilkie Professor Stephen Withers SECTIONAL COMMITTEE 2 [1963] SECTIONAL COMMITTEE 4 [1990] Astronomy and physics Engineering Chair: Professor Simon White Chair: Professor Hywel Thomas Members: Members: Professor Girish Agarwal Professor Ross Anderson Professor Michael Coey Professor Alan Bundy Professor Jack Connor Professor Michael Burdekin Professor Laurence Eaves Professor Russell Cowburn Professor Nigel Glover Professor John Crowcroft